|
受注:045-509-1970 |
技術サポート:[email protected] 平日9:00〜18:00 1営業日以内にご連絡を差し上げます |
化学情報
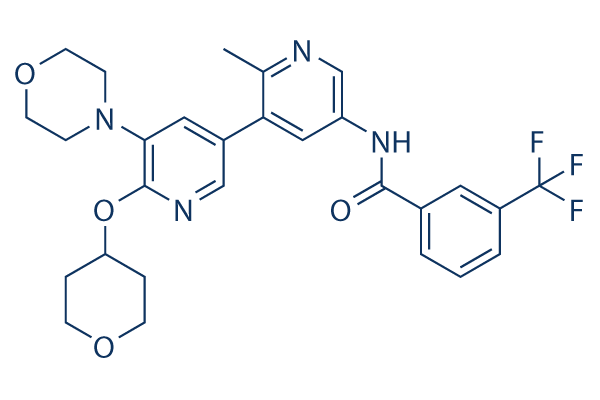
|
Synonyms | N/A | Storage (From the date of receipt) |
3 years -20°C powder 1 years -80°C in solvent |
| 化学式 | C28H29F3N4O4 |
|||
| 分子量 | 542.55 | CAS No. | 1628838-42-5 | |
| Solubility (25°C)* | 体外 | DMSO | 100 mg/mL (184.31 mM) | |
| Ethanol | 100 mg/mL (184.31 mM) | |||
| Water | Insoluble | |||
|
* <1 mg/ml means slightly soluble or insoluble. * Please note that Selleck tests the solubility of all compounds in-house, and the actual solubility may differ slightly from published values. This is normal and is due to slight batch-to-batch variations. |
||||
溶剤液(一定の濃度)を調合する
生物活性
| 製品説明 | RAF709 is a potent inhibitor of B/C RAF kinase with almost equivalent IC50 values of 0.4 nM for B-RAF and C-RAF, showing a high level of selectivity, demonstrating greater than 99% on-target binding to BRAF, BRAFV600E, and CRAF at 1 μM and very few off-targets with DDR1 (>99%), DDR2 (86%), FRK (92%), and PDGFRb (96%), the only kinases with binding >80% at 1 μM. |
|---|---|
| in vitro | RAF709 appears to have very slow dissociation kinetics (T1/2 > 6.5 h) using the rapid dilution method to measure its dissociation rate constant. In cellular assays, the dose−response of pMEK and pERK are measured in Calu-6 cells with EC50 of 0.02 and 0.1 μM with minimal paradoxical activation and inhibition of proliferation with EC50 of 0.95 μM. RAF709 stabilizes BRAF−CRAF dimers with an EC50 of 0.8 μM. Of the 456 kinases tested, RAF709 shows a high level of selectivity, demonstrating greater than 99% on-target binding to BRAF, BRAFV600E, and CRAF at 1 μM and very few off-targets with DDR1 (>99%), DDR2 (86%), FRK (92%), and PDGFRb (96%), the only kinases with binding >80% at 1 μM[1]. RAF709 shows equal activity against both RAF monomers and dimers. In in vitro biochemical assays, RAF709 exhibits potent inhibitory activity targeting BRAF, BRAFV600E, and CRAF with IC50 values ranging between 0.3 to 1.5 nmol/L. RAF709 treatment leads to a dose-dependent induction of B/CRAF heterodimerization in HCT116, but inhibits MEK and ERK phosphorylation, in line with the ability of RAF709 to effectively inhibit the RAF dimers. RAF709 selectively inhibits oncogenic signaling and proliferation in tumor cells with BRAF, NRAS, or KRAS mutations with minimal paradoxical activation[2]. |
| in vivo | RAF709 is well tolerated and efficacious in KRAS mutant xenograft models. It is reasonably stable in plasma after a 3 h incubation at 37℃ across species [plasma stability (%remaining): rat 85%, mouse 82%, dog 95%, human 101%], and plasma protein binding is measured to be 98% across species. In pharmacokinetic experiments, RAF709 has moderate clearance in mouse (35 mL/min/kg) and dog (14 mL/min/kg) and high clearance in rat (50 mL/min/kg). Cmax in mouse (1 μM), dog (0.5 μM), and rat (0.5 μM) reach pharmacologically active concentrations, and acceptable oral availability is observed in mouse (68%), rat (24%), and dog (48%). In the Calu-6 xenograft nude mouse model, treatment with RAF709 results in dose-dependent antitumor activity with 10 mg/kg being subefficacious (%T/C = 92%), 30 mg/kg resulted in measurable antitumor activity (% T/C = 46%), and 200 mg/kg resulted in mean tumor regression of 92%, while the same high dose is not efficacious in the PC3, KRAS WT mode[1]. |
プロトコル(参考用のみ)
| キナーゼアッセイ | CRAF kinase assay | |
|---|---|---|
| The CRAF kinase assay was carried out using 10 nM kinase-dead MEK1 protein substrate (carrying a K97R mutation), 3 μM ATP, and 10 pM CRAF Y340E/Y341E. The reaction buffer contained 50 mM Tris pH 7.5, 10 mM MgCl2, 0.05% BSA, 50 mM NaCl, 0.01% Tween-20, and 1 mM DTT. The reactions were carried out at room temperature in a volume of 10 μL in white 384-shallow-well plates for 40 min and stopped by adding 5 μL/well quench solution (50 mM Tris pH 7.5, 50 mM EDTA). Terminated reactions received 5 μL/well detection reagents consisting of 50 mM Tris pH 7.5, 0.01% Tween-20, 1:1000 diluted antiphospho MEK1/2 S217/S221 antibody, 0.01 mg/mL each of AlphaScreen Protein A-coated acceptor beads, and streptavidin-coated donor bead. Plates were read in an EnVision plate reader after overnight incubation at room temperature. In compound inhibition studies, compounds were tested over a concentration range of 25 μM to 1.74 × 10−6 μM in 16-point, 3-fold format. DMSO was at a final concentration of 0.5%. Compounds were preincubated with CRAF for 30 min before adding substrates to start the reaction. Inhibition data were fit to a four-parameter logistic equation to calculate the IC50 of the compounds. | ||
| 細胞アッセイ | 細胞株 | HCT116 cells |
| 濃度 | 0.1, 1, 10 μM | |
| 反応時間 | 1 hour | |
| 実験の流れ | HCT116 cells are treated with DMSO or RAF709 at indicated concentrations for 1 hour; 1 mmol/L dabrafenib treatment is included for comparison. BRAF/CRAF dimerization is assessed by immunoprecipitating BRAF or CRAF, followed by Western blot analysis of BRAF and CRAF. Levels of pMEK and pERK in whole-cell lysates (WCL) are determined by Western blot analysis. GAPDH level is included as a loading control. |
|
| 動物実験 | 動物モデル | Calu-6 model (tumor bearing mice) |
| 投薬量 | 10, 30, or 200 mg/kg | |
| 投与方法 | oral administration | |
参考
|
Selleckの高級品が、幾つかの出版された研究調査結果(以下を含む)で使われた:
| Identification of a RAS-activating TMEM87A-RASGRF1 fusion in an exceptional responder to sunitinib with non-small cell lung cancer. [ Clin Cancer Res, 2020, 20 pii: clincanres] | PubMed: 32312893 |
| KRASQ61H preferentially signals through MAPK in a RAF dimer-dependent manner in non-small cell lung cancer [ Cancer Res, 2020, canres.0448.2020] | PubMed: 32605999 |
| Depolarization-Dependent C-Raf Signaling Promotes Hyperexcitability and Reduces Opioid Sensitivity of Isolated Nociceptors after Spinal Cord Injury [ J Neurosci, 2020, 40(34):6522-6535] | PubMed: 32690613 |
長期の保管のために-20°Cの下で製品を保ってください。
人間や獣医の診断であるか治療的な使用のためにでない。
各々の製品のための特定の保管と取扱い情報は、製品データシートの上で示されます。大部分のSelleck製品は、推薦された状況の下で安定です。製品は、推薦された保管温度と異なる温度で、時々出荷されます。長期の保管のために必要とされてそれと異なる温度で、多くの製品は、短期もので安定です。品質を維持するが、夜通しの積荷のために最も経済的な貯蔵状況を用いてあなたの送料を保存する状況の下に、製品が出荷されることを、我々は確実とします。製品の受領と同時に、製品データシートの上で貯蔵推薦に従ってください。