|
受注:045-509-1970 |
技術サポート:[email protected] 平日9:00〜18:00 1営業日以内にご連絡を差し上げます |
化学情報
|
Synonyms | Fxr 450 | Storage (From the date of receipt) |
3 years -20°C powder 1 years -80°C in solvent |
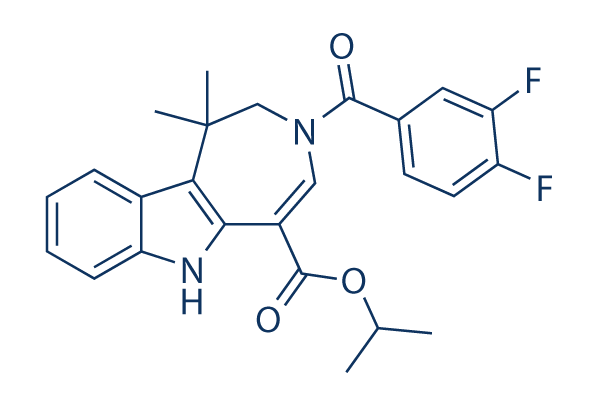
| 化学式 | C25H24F2N2O3 |
|||
| 分子量 | 438.47 | CAS No. | 629664-81-9 | |
| Solubility (25°C)* | 体外 | DMSO | 33 mg/mL warmed with 50ºC water bath (75.26 mM) | |
| Ethanol | 2 mg/mL (4.56 mM) | |||
| Water | Insoluble | |||
|
* <1 mg/ml means slightly soluble or insoluble. * Please note that Selleck tests the solubility of all compounds in-house, and the actual solubility may differ slightly from published values. This is normal and is due to slight batch-to-batch variations. |
||||
溶剤液(一定の濃度)を調合する
生物活性
| 製品説明 | Turofexorate Isopropyl (XL335, Fxr 450) is a potent, selective FXR agonist with EC50 of 4 nM, highly selective versus other nuclear receptors, such as LXR, PPAR, ER and etc. Phase 1. |
|---|---|
| in vitro | WAY-362450 binds to the ligand-binding domain (LBD) of human FXR. WAY-362450 resides in a predominately hydrophobic pocket with only a few polar atoms making contact with WAY-362450. WAY-362450 promotes transcription of the human BSEP, human SHP, and mouse IBABP genes utilizing reporter constructs with EC50 of 17, 230, and 33 nM, respectively in promoter assays. WAY-362450 at concentration of 1 μM significantly induces mRNAs encoding for BSEP, SHP, and IBABP in human cell cultures to 13-, 2-, and 20-fold, respectively. [1] WAY-362450 at concentration of 1 μM suppresses interleukin-6-induced CRP expression in human Hep3B hepatoma cells, and the inhibitory effect is attenuated when knockdown of FXR by short interfering RNA. |
| in vivo | WAY-362450 administrated intravenously or orally at does of 3 mg/kg in rats with a protracted half-life of 25 h, modest volume of distribution, and low clearance of 3.3 L/kg. WAY-362450 administered orally at dose of 10 mg/kg in normal C57bl/6 mice for a period of 7 days significantly lowers triglycerides to 62.0 ± 6.4 mg/dL and total cholesterol to 78.1 ± 5.0 mg/dL. WAY-362450 administered orally at dose of 1 and 3 mg/kg daily for 6 weeks in LDLR−/− mice, triglycerides is lowered by 19% and 39%, respectively, total cholesterol is lowered by 23% and 50%, respectively and lesion formation by 18% and 36%, respectively. [1] WAY-362450 intraperitoneally administrated at dose of 30 mg/kg daily for 4 days in wild type C57BL/6 mice attenuates lipopolysaccharide-induced serum amyloid P component and serum amyloid A3 mRNA levels in the liver. [3] WAY-362450 orally administered at dose of 30 mg/kg/day for 4 weeks in adult male C57BL/6 mice reduces inflammatory cell infiltration and hepatic fibrosis, the reduction in inflammatory cell infiltration correlates with deceased serum levels of keratinocyte derived chemokine (mKC) and MCP 1 and decreased hepatic gene expression of MCP-1 and VCAM-1, and the reduction of hepatic fibrosis by WAY-362450 treatment corresponded to a reduction in hepatic gene expression of fibrosis markers. [3] WAY-362450 administrated orally at dose of 30mg/kg in LDLR−/− and apoE−/− mice blocks diet-induced hypertriglyceridemia and elevations of non-HDL cholesterol and produced a near complete inhibition of aortic lesion formation, WAY-362450 also induced small heterodimer partner (SHP) expression and repressed cholesterol 7α-hydroxylase (CYP7A1) and sterol 12 α-hydroxylase (CYP8B1) expression. [4] |
プロトコル(参考用のみ)
| キナーゼアッセイ | Cross-reactivity Studies | |
|---|---|---|
| The selectivity of 6m is evaluated using a panel of nuclear receptor expression plasmids in the forms of wild type receptors or chimeric receptors with Gal-DBD and NHR LBD transiently transfected to CV-1 cells in tissue culture flasks with the NHR responsive luciferase reporters. Except VDR assay, it is a two hybrid assay that measure the interaction of VP16 fused VDR-LBD and Gal-SRC1. The cells are harvested after 6 h transfection and seeded to the assay plates with diluted testing WAY-362450. Different than the agonist assay, the antagonist assay is carried out the in presence of an agonist at concentration of EC75. After 18 h of incubation, the media is removed from the plates and the lucifaerase activity is determined using the luciferase substrate. As shown in Tables S4-S5, compound 6m is highly selective, as little or no significant cross-reactivity with other nuclear hormone receptors is observed at concentrations up to 10 μM | ||
| 動物実験 | 動物モデル | Seven week old male C57bl/6 mice |
| 投薬量 | 10 mg/kg | |
| 投与方法 | Administrated orally once daily in the morning for a period of 7 days | |
参考
カスタマーフィードバック
-
, , Oncotarget, 2015, 6(6):4226-38.
Selleckの高級品が、幾つかの出版された研究調査結果(以下を含む)で使われた:
| Chronic activation of FXR-induced liver growth with tissue-specific targeting Cyclin D1. [ Cell Cycle, 2019, 18(15):1784-1797] | PubMed: 31223053 |
| Farnesoid X receptor associates with β-catenin and inhibits its activity in hepatocellular carcinoma [Liu X, et al. Oncotarget, 2015, 6(6):4226-38] | PubMed: 25650661 |
| Agonist of farnesoid X receptor protects against bile acid induced damage and oxidative stress in mouse placenta--a study on maternal cholestasis model. [ Placenta, 2015, 36(5):545-51] | PubMed: 25747729 |
| Activation of farnesoid X receptor induces RECK expression in mouse liver. [ Biochem Biophys Res Commun, 2014, 443(1):211-6] | PubMed: 24291500 |
長期の保管のために-20°Cの下で製品を保ってください。
人間や獣医の診断であるか治療的な使用のためにでない。
各々の製品のための特定の保管と取扱い情報は、製品データシートの上で示されます。大部分のSelleck製品は、推薦された状況の下で安定です。製品は、推薦された保管温度と異なる温度で、時々出荷されます。長期の保管のために必要とされてそれと異なる温度で、多くの製品は、短期もので安定です。品質を維持するが、夜通しの積荷のために最も経済的な貯蔵状況を用いてあなたの送料を保存する状況の下に、製品が出荷されることを、我々は確実とします。製品の受領と同時に、製品データシートの上で貯蔵推薦に従ってください。