
- 阻害剤
- 研究分野別
- PI3K/Akt/mTOR
- Epigenetics
- Methylation
- Immunology & Inflammation
- Protein Tyrosine Kinase
- Angiogenesis
- Apoptosis
- Autophagy
- ER stress & UPR
- JAK/STAT
- MAPK
- Cytoskeletal Signaling
- Cell Cycle
- TGF-beta/Smad
- 化合物ライブラリー
- 抗体
- 新製品
- お問い合わせ
Sorafenib (BAY 43-9006)
別名:NSC-724772,BAY 43-9006
Sorafenib is a multikinase inhibitor of Raf-1 and B-Raf with IC50 of 6 nM and 22 nM in cell-free assays, respectively. Sorafenib inhibits VEGFR-2, VEGFR-3, PDGFR-β, Flt-3 and c-KIT with IC50 of 90 nM, 20 nM, 57 nM, 59 nM and 68 nM, respectively. Sorafenib induces autophagy and apoptosis and activates ferroptosis with anti-tumor activity.
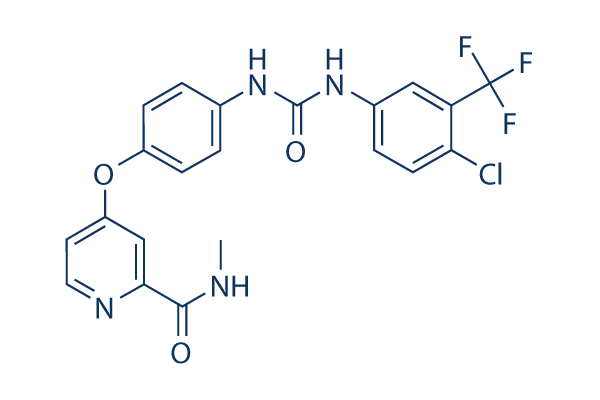
CAS No. 284461-73-0
文献中Selleckの製品使用例(580)
製品安全説明書
現在のバッチを見る:
純度:
99.91%
99.91
Sorafenib (BAY 43-9006)関連製品
シグナル伝達経路
Raf阻害剤の選択性比較
Cell Data
| Cell Lines | Assay Type | Concentration | Incubation Time | 活性情報 | PMID |
|---|---|---|---|---|---|
| OS-RC-2 | Growth Inhibition Assay | IC50=9.11243 μM | SANGER | ||
| LC-1F | Growth Inhibition Assay | IC50=9.10834 μM | SANGER | ||
| TE-10 | Growth Inhibition Assay | IC50=8.76353 μM | SANGER | ||
| HD-MY-Z | Growth Inhibition Assay | IC50=8.63746 μM | SANGER | ||
| NB14 | Growth Inhibition Assay | IC50=8.63133 μM | SANGER | ||
| IST-MEL1 | Growth Inhibition Assay | IC50=8.40002 μM | SANGER | ||
| RPMI-8226 | Growth Inhibition Assay | IC50=7.84511 μM | SANGER | ||
| ES8 | Growth Inhibition Assay | IC50=7.76503 μM | SANGER | ||
| D-392MG | Growth Inhibition Assay | IC50=7.62663 μM | SANGER | ||
| NB1 | Growth Inhibition Assay | IC50=7.40407 μM | SANGER | ||
| 697 | Growth Inhibition Assay | IC50=7.05989 μM | SANGER | ||
| DOHH-2 | Growth Inhibition Assay | IC50=7.05676 μM | SANGER | ||
| SIMA | Growth Inhibition Assay | IC50=7.00101 μM | SANGER | ||
| OCI-AML2 | Growth Inhibition Assay | IC50=6.61062 μM | SANGER | ||
| ML-2 | Growth Inhibition Assay | IC50=6.52849 μM | SANGER | ||
| SW954 | Growth Inhibition Assay | IC50=6.45866 μM | SANGER | ||
| MZ2-MEL | Growth Inhibition Assay | IC50=6.31839 μM | SANGER | ||
| MOLT-16 | Growth Inhibition Assay | IC50=6.25266 μM | SANGER | ||
| SR | Growth Inhibition Assay | IC50=6.00678 μM | SANGER | ||
| BV-173 | Growth Inhibition Assay | IC50=5.95682 μM | SANGER | ||
| no-11 | Growth Inhibition Assay | IC50=5.73568 μM | SANGER | ||
| JAR | Growth Inhibition Assay | IC50=5.50824 μM | SANGER | ||
| NCI-H510A | Growth Inhibition Assay | IC50=5.41685 μM | SANGER | ||
| TE-12 | Growth Inhibition Assay | IC50=5.24929 μM | SANGER | ||
| SF539 | Growth Inhibition Assay | IC50=5.13227 μM | SANGER | ||
| QIMR-WIL | Growth Inhibition Assay | IC50=5.07294 μM | SANGER | ||
| NOMO-1 | Growth Inhibition Assay | IC50=4.48905 μM | SANGER | ||
| MHH-PREB-1 | Growth Inhibition Assay | IC50=4.40484 μM | SANGER | ||
| NCI-H1581 | Growth Inhibition Assay | IC50=4.28798 μM | SANGER | ||
| BC-3 | Growth Inhibition Assay | IC50=4.23391 μM | SANGER | ||
| SK-NEP-1 | Growth Inhibition Assay | IC50=4.16815 μM | SANGER | ||
| KS-1 | Growth Inhibition Assay | IC50=3.88198 μM | SANGER | ||
| ST486 | Growth Inhibition Assay | IC50=3.83673 μM | SANGER | ||
| BC-1 | Growth Inhibition Assay | IC50=3.7402 μM | SANGER | ||
| MZ7-mel | Growth Inhibition Assay | IC50=3.66099 μM | SANGER | ||
| MRK-nu-1 | Growth Inhibition Assay | IC50=3.61468 μM | SANGER | ||
| MZ1-PC | Growth Inhibition Assay | IC50=3.47509 μM | SANGER | ||
| TE-5 | Growth Inhibition Assay | IC50=3.13162 μM | SANGER | ||
| HCC2218 | Growth Inhibition Assay | IC50=3.12003 μM | SANGER | ||
| HL-60 | Growth Inhibition Assay | IC50=3.06299 μM | SANGER | ||
| MFH-ino | Growth Inhibition Assay | IC50=2.92185 μM | SANGER | ||
| KMOE-2 | Growth Inhibition Assay | IC50=2.8135 μM | SANGER | ||
| L-540 | Growth Inhibition Assay | IC50=2.75789 μM | SANGER | ||
| K052 | Growth Inhibition Assay | IC50=2.74616 μM | SANGER | ||
| BE-13 | Growth Inhibition Assay | IC50=2.69609 μM | SANGER | ||
| SIG-M5 | Growth Inhibition Assay | IC50=2.42242 μM | SANGER | ||
| NCI-H2171 | Growth Inhibition Assay | IC50=2.39764 μM | SANGER | ||
| NCI-SNU-1 | Growth Inhibition Assay | IC50=2.3162 μM | SANGER | ||
| OVCAR-4 | Growth Inhibition Assay | IC50=2.21393 μM | SANGER | ||
| D-247MG | Growth Inhibition Assay | IC50=2.1448 μM | SANGER | ||
| A3-KAW | Growth Inhibition Assay | IC50=1.8842 μM | SANGER | ||
| MEG-01 | Growth Inhibition Assay | IC50=1.8098 μM | SANGER | ||
| SF126 | Growth Inhibition Assay | IC50=1.63812 μM | SANGER | ||
| no-10 | Growth Inhibition Assay | IC50=1.61726 μM | SANGER | ||
| A388 | Growth Inhibition Assay | IC50=1.59165 μM | SANGER | ||
| KG-1 | Growth Inhibition Assay | IC50=1.47935 μM | SANGER | ||
| LAMA-84 | Growth Inhibition Assay | IC50=1.37648 μM | SANGER | ||
| EM-2 | Growth Inhibition Assay | IC50=1.35578 μM | SANGER | ||
| NCI-H1436 | Growth Inhibition Assay | IC50=1.21678 μM | SANGER | ||
| LB2518-MEL | Growth Inhibition Assay | IC50=1.20809 μM | SANGER | ||
| KM12 | Growth Inhibition Assay | IC50=1.02098 μM | SANGER | ||
| GDM-1 | Growth Inhibition Assay | IC50=0.90698 μM | SANGER | ||
| HCC2998 | Growth Inhibition Assay | IC50=0.88818 μM | SANGER | ||
| HOP-62 | Growth Inhibition Assay | IC50=0.85088 μM | SANGER | ||
| SH-4 | Growth Inhibition Assay | IC50=0.65613 μM | SANGER | ||
| NOS-1 | Growth Inhibition Assay | IC50=0.5636 μM | SANGER | ||
| BB65-RCC | Growth Inhibition Assay | IC50=0.47073 μM | SANGER | ||
| CGTH-W-1 | Growth Inhibition Assay | IC50=0.25022 μM | SANGER | ||
| NKM-1 | Growth Inhibition Assay | IC50=0.07416 μM | SANGER | ||
| ALL-PO | Growth Inhibition Assay | IC50=0.03184 μM | SANGER | ||
| MONO-MAC-6 | Growth Inhibition Assay | IC50=0.00418 μM | SANGER | ||
| MV-4-11 | Growth Inhibition Assay | IC50=0.00000303 μM | SANGER | ||
| NCI-SNU-16 | Growth Inhibition Assay | IC50=9.21026 μM | SANGER | ||
| SHP-77 | Growth Inhibition Assay | IC50=9.71662 μM | SANGER | ||
| A4-Fuk | Growth Inhibition Assay | IC50=9.7561 μM | SANGER | ||
| NB6 | Growth Inhibition Assay | IC50=9.76029 μM | SANGER | ||
| JiyoyeP-2003 | Growth Inhibition Assay | IC50=10.4745 μM | SANGER | ||
| DMS-114 | Growth Inhibition Assay | IC50=10.5441 μM | SANGER | ||
| NB7 | Growth Inhibition Assay | IC50=10.7526 μM | SANGER | ||
| NCI-H747 | Growth Inhibition Assay | IC50=11.1216 μM | SANGER | ||
| HH | Growth Inhibition Assay | IC50=11.3876 μM | SANGER | ||
| EW-18 | Growth Inhibition Assay | IC50=11.9044 μM | SANGER | ||
| CHP-126 | Growth Inhibition Assay | IC50=11.9738 μM | SANGER | ||
| NTERA-S-cl-D1 | Growth Inhibition Assay | IC50=12.0278 μM | SANGER | ||
| DEL | Growth Inhibition Assay | IC50=12.0985 μM | SANGER | ||
| LU-139 | Growth Inhibition Assay | IC50=12.5413 μM | SANGER | ||
| P30-OHK | Growth Inhibition Assay | IC50=12.5479 μM | SANGER | ||
| NCI-H1522 | Growth Inhibition Assay | IC50=12.746 μM | SANGER | ||
| NCI-H1299 | Growth Inhibition Assay | IC50=13.2911 μM | SANGER | ||
| UACC-257 | Growth Inhibition Assay | IC50=13.5126 μM | SANGER | ||
| Calu-6 | Growth Inhibition Assay | IC50=13.6046 μM | SANGER | ||
| NCI-H1882 | Growth Inhibition Assay | IC50=13.8555 μM | SANGER | ||
| BB30-HNC | Growth Inhibition Assay | IC50=14.0609 μM | SANGER | ||
| ES1 | Growth Inhibition Assay | IC50=14.1551 μM | SANGER | ||
| NCI-H1694 | Growth Inhibition Assay | IC50=14.4811 μM | SANGER | ||
| IST-SL1 | Growth Inhibition Assay | IC50=14.9616 μM | SANGER | ||
| ECC4 | Growth Inhibition Assay | IC50=15.0558 μM | SANGER | ||
| MDA-MB-134-VI | Growth Inhibition Assay | IC50=15.4131 μM | SANGER | ||
| SCH | Growth Inhibition Assay | IC50=15.4728 μM | SANGER | ||
| SK-N-FI | Growth Inhibition Assay | IC50=15.6534 μM | SANGER | ||
| HDLM-2 | Growth Inhibition Assay | IC50=16.0714 μM | SANGER | ||
| Ramos-2G6-4C10 | Growth Inhibition Assay | IC50=16.1297 μM | SANGER | ||
| EW-24 | Growth Inhibition Assay | IC50=16.1661 μM | SANGER | ||
| NCI-H2141 | Growth Inhibition Assay | IC50=16.189 μM | SANGER | ||
| LC4-1 | Growth Inhibition Assay | IC50=16.6119 μM | SANGER | ||
| HT-144 | Growth Inhibition Assay | IC50=17.006 μM | SANGER | ||
| SK-MEL-1 | Growth Inhibition Assay | IC50=17.0072 μM | SANGER | ||
| SCC-15 | Growth Inhibition Assay | IC50=17.1638 μM | SANGER | ||
| C8166 | Growth Inhibition Assay | IC50=17.6833 μM | SANGER | ||
| GOTO | Growth Inhibition Assay | IC50=17.8344 μM | SANGER | ||
| COR-L279 | Growth Inhibition Assay | IC50=18.1362 μM | SANGER | ||
| K-562 | Growth Inhibition Assay | IC50=18.7143 μM | SANGER | ||
| ES3 | Growth Inhibition Assay | IC50=18.8041 μM | SANGER | ||
| LU-165 | Growth Inhibition Assay | IC50=19.7008 μM | SANGER | ||
| KM-H2 | Growth Inhibition Assay | IC50=20.3184 μM | SANGER | ||
| RL | Growth Inhibition Assay | IC50=20.9692 μM | SANGER | ||
| EW-3 | Growth Inhibition Assay | IC50=21.1889 μM | SANGER | ||
| A101D | Growth Inhibition Assay | IC50=21.3752 μM | SANGER | ||
| HUTU-80 | Growth Inhibition Assay | IC50=21.3946 μM | SANGER | ||
| NCI-H23 | Growth Inhibition Assay | IC50=21.3992 μM | SANGER | ||
| PF-382 | Growth Inhibition Assay | IC50=21.4403 μM | SANGER | ||
| LB373-MEL-D | Growth Inhibition Assay | IC50=21.5615 μM | SANGER | ||
| TE-8 | Growth Inhibition Assay | IC50=21.6394 μM | SANGER | ||
| TE-9 | Growth Inhibition Assay | IC50=21.8513 μM | SANGER | ||
| Daudi | Growth Inhibition Assay | IC50=21.9304 μM | SANGER | ||
| D-542MG | Growth Inhibition Assay | IC50=22.0256 μM | SANGER | ||
| U-698-M | Growth Inhibition Assay | IC50=22.4603 μM | SANGER | ||
| ES6 | Growth Inhibition Assay | IC50=22.7366 μM | SANGER | ||
| DU-4475 | Growth Inhibition Assay | IC50=23.8897 μM | SANGER | ||
| ECC12 | Growth Inhibition Assay | IC50=24.2803 μM | SANGER | ||
| C2BBe1 | Growth Inhibition Assay | IC50=24.3239 μM | SANGER | ||
| IST-SL2 | Growth Inhibition Assay | IC50=24.4362 μM | SANGER | ||
| DJM-1 | Growth Inhibition Assay | IC50=24.5221 μM | SANGER | ||
| DMS-153 | Growth Inhibition Assay | IC50=24.8614 μM | SANGER | ||
| NB13 | Growth Inhibition Assay | IC50=25.0265 μM | SANGER | ||
| SK-N-DZ | Growth Inhibition Assay | IC50=26.3414 μM | SANGER | ||
| COR-L88 | Growth Inhibition Assay | IC50=26.5796 μM | SANGER | ||
| LU-65 | Growth Inhibition Assay | IC50=26.8535 μM | SANGER | ||
| TGBC1TKB | Growth Inhibition Assay | IC50=26.9828 μM | SANGER | ||
| THP-1 | Growth Inhibition Assay | IC50=27.2141 μM | SANGER | ||
| ONS-76 | Growth Inhibition Assay | IC50=27.332 μM | SANGER | ||
| LC-2-ad | Growth Inhibition Assay | IC50=27.6231 μM | SANGER | ||
| EW-13 | Growth Inhibition Assay | IC50=29.1746 μM | SANGER | ||
| MS-1 | Growth Inhibition Assay | IC50=30.7278 μM | SANGER | ||
| NCI-H2227 | Growth Inhibition Assay | IC50=30.9806 μM | SANGER | ||
| LXF-289 | Growth Inhibition Assay | IC50=31.4492 μM | SANGER | ||
| MC116 | Growth Inhibition Assay | IC50=32.0826 μM | SANGER | ||
| EVSA-T | Growth Inhibition Assay | IC50=32.2585 μM | SANGER | ||
| CTB-1 | Growth Inhibition Assay | IC50=33.1101 μM | SANGER | ||
| COLO-320-HSR | Growth Inhibition Assay | IC50=33.1603 μM | SANGER | ||
| NCI-H2196 | Growth Inhibition Assay | IC50=33.2557 μM | SANGER | ||
| LB2241-RCC | Growth Inhibition Assay | IC50=33.3135 μM | SANGER | ||
| LS-513 | Growth Inhibition Assay | IC50=33.8638 μM | SANGER | ||
| LP-1 | Growth Inhibition Assay | IC50=33.9956 μM | SANGER | ||
| A253 | Growth Inhibition Assay | IC50=34.2296 μM | SANGER | ||
| SK-MM-2 | Growth Inhibition Assay | IC50=34.9451 μM | SANGER | ||
| NCI-H1963 | Growth Inhibition Assay | IC50=35.3072 μM | SANGER | ||
| MMAC-SF | Growth Inhibition Assay | IC50=35.8785 μM | SANGER | ||
| LB831-BLC | Growth Inhibition Assay | IC50=36.0654 μM | SANGER | ||
| WSU-NHL | Growth Inhibition Assay | IC50=36.164 μM | SANGER | ||
| CESS | Growth Inhibition Assay | IC50=36.2848 μM | SANGER | ||
| NEC8 | Growth Inhibition Assay | IC50=36.5835 μM | SANGER | ||
| KNS-42 | Growth Inhibition Assay | IC50=37.1237 μM | SANGER | ||
| MHH-CALL-2 | Growth Inhibition Assay | IC50=37.1821 μM | SANGER | ||
| K5 | Growth Inhibition Assay | IC50=38.43 μM | SANGER | ||
| CP66-MEL | Growth Inhibition Assay | IC50=39.0733 μM | SANGER | ||
| OPM-2 | Growth Inhibition Assay | IC50=39.8432 μM | SANGER | ||
| IST-MES1 | Growth Inhibition Assay | IC50=40.3096 μM | SANGER | ||
| EC-GI-10 | Growth Inhibition Assay | IC50=41.5805 μM | SANGER | ||
| CTV-1 | Growth Inhibition Assay | IC50=42.8406 μM | SANGER | ||
| DG-75 | Growth Inhibition Assay | IC50=43.7595 μM | SANGER | ||
| KNS-81-FD | Growth Inhibition Assay | IC50=45.4058 μM | SANGER | ||
| NCI-H82 | Growth Inhibition Assay | IC50=45.5758 μM | SANGER | ||
| RPMI-8866 | Growth Inhibition Assay | IC50=46.1873 μM | SANGER | ||
| ACN | Growth Inhibition Assay | IC50=46.434 μM | SANGER | ||
| NCI-H1395 | Growth Inhibition Assay | IC50=46.4756 μM | SANGER | ||
| NCI-H209 | Growth Inhibition Assay | IC50=47.1405 μM | SANGER | ||
| TGW | Growth Inhibition Assay | IC50=49.0791 μM | SANGER | ||
| NCI-H748 | Growth Inhibition Assay | IC50=49.4753 μM | SANGER | ||
| EKVX | Growth Inhibition Assay | IC50=49.6628 μM | SANGER | ||
| MV4-11 | Antiproliferative activity assay | IC50 = 0.00087 μM | 19754199 | ||
| MV4-11 | Cytotoxicity assay | IC50 = 0.00087 μM | 19654408 | ||
| TT | Antiproliferative activity assay | IC50 = 0.0014 μM | 18849971 | ||
| RS4-11 | Function assay | IC50 = 0.002 μM | 19654408 | ||
| MOLM13 | Function assay | IC50 = 0.002 μM | 24641103 | ||
| Sf9 | Function assay | IC50 = 0.003 μM | 21708468 | ||
| RS4-11 | Function assay | IC50 = 0.0032 μM | 19654408 | ||
| MV4-11 | Growth inhibition assay | IC50 = 0.004 μM | 29357250 | ||
| A375 | Function assay | IC50 = 0.0044 μM | 29461827 | ||
| MOLM13 | Antiproliferative activity assay | IC50 = 0.005 μM | 24641103 | ||
| MV4-11 | Function assay | IC50 = 0.007 μM | 23362959 | ||
| MV4-11 | Cytotoxicity assay | IC50 = 0.007 μM | 26342867 | ||
| HEK293 | Function assay | Kd = 0.013 μM | 19754199 | ||
| Kasumi-1 | Antiproliferative activity assay | IC50 = 0.015 μM | 20570526 | ||
| MV4-11 | Antiproliferative activity assay | IC50 = 0.015 μM | 20570526 | ||
| sf9 | Function assay | IC50 = 0.015 μM | 29266937 | ||
| sf9 | Function assay | IC50 = 0.018 μM | 21708468 | ||
| sf9 | Function assay | IC50 = 0.02 μM | 23618709 | ||
| MV4-11 | Antiproliferative activity assay | GI50 = 0.03 μM | 21708468 | ||
| Sf9 | Function assay | IC50 = 0.042 μM | 26081023 | ||
| SF9 | Function assay | IC50 = 0.043 μM | 18473434 | ||
| MV4-11 | Cytotoxicity assay | GI50 = 0.043 μM | 23618709 | ||
| MV4-11 | Function assay | GI50 = 0.043 μM | 26081023 | ||
| SF9 | Function assay | IC50 = 0.044 μM | 26081023 | ||
| SF9 | Function assay | IC50 = 0.054 μM | 21708468 | ||
| SF9 | Function assay | IC50 = 0.054 μM | 23618709 | ||
| MOLM13 | Antiproliferative activity assay | GI50 = 0.056 μM | 21708468 | ||
| MOLM13 | Antiproliferative activity assay | GI50 = 0.056 μM | 22726931 | ||
| MOLM13 | Cytotoxicity assay | GI50 = 0.056 μM | 23618709 | ||
| MOLM-13 | Function assay | GI50 = 0.056 μM | 26081023 | ||
| SF9 | Function assay | IC50 = 0.058 μM | 29266937 | ||
| WM266.4 | Antiproliferative activity assay | IC50 = 0.06 μM | 25496804 | ||
| MDA-MB-231 | Function assay | IC50 = 0.063 μM | 28431342 | ||
| SF9 | Function assay | IC50 = 0.09 μM | 29266937 | ||
| Spodoptera frugiperda | Function assay | IC50 = 0.09 μM | 26922228 | ||
| SF9 | Function assay | IC50 = 0.09 μM | 23562241 | ||
| SF9 | Function assay | IC50 = 0.1 μM | 23442188 | ||
| SF9 | Function assay | IC50 = 0.1 μM | 23442188 | ||
| HEK293 | Function assay | IC50 = 0.12 μM | 26318998 | ||
| MCF7 | Antiproliferative activity assay | IC50 = 0.19 μM | 25496804 | ||
| MV4-11 | Antiproliferative activity human | IC50 = 0.3 μM | 28242553 | ||
| HepG2 | Antiproliferative activity assay | EC50 = 0.302 μM | 27010810 | ||
| THP1 | Antiproliferative activity assay | IC50 = 0.31 μM | 20570526 | ||
| SMMC7721 | Cytotoxicity assay | IC50 = 0.37 μM | 25982075 | ||
| HT-29 | Antiproliferative activity assay | IC50 = 0.39 μM | 25778995 | ||
| sf9 | Function assay | Kd = 0.6 μM | 28109791 | ||
| T47D | Antiproliferative activity assay | IC50 = 0.61 μM | 25778995 | ||
| A549 | Cytotoxicity assay | IC50 = 0.63 μM | 25982075 | ||
| HeLa | Cytotoxicity assay | IC50 = 0.64 μM | 30015070 | ||
| WM3629 | Antiproliferative activity assay | GI50 = 0.65 μM | 20466542 | ||
| SMMC7721 | Antiproliferative activity assay | IC50 = 0.65 μM | 26342134 | ||
| WM3629 | Growth inhibition assay | GI50 = 0.78 μM | 20149658 | ||
| WM3629 | Antiproliferative activity assay | GI50 = 0.78 μM | 26810260 | ||
| MCF7 | Antiproliferative activity assay | IC50 = 0.78 μM | 25778995 | ||
| K562 | Antiproliferative activity assay | IC50 = 0.86 μM | 24315192 | ||
| COLO205 | Antiproliferative activity assay | IC50 = 0.87 μM | 25778995 | ||
| T29 | Cytotoxicity assay | IC50 = 0.9 μM | 26318998 | ||
| MDA-MB-231 | Cytotoxicity assay | IC50 = 0.94 μM | 25086238 | ||
| MDA-MB-231 | Cytotoxicity assay | IC50 = 0.94 μM | 25440879 | ||
| HepG2 | Antiproliferative activity assay | IC50 = 1.06 μM | 29628325 | ||
| BL21(DE3) | Function assay | IC50 = 1.1 μM | 19928858 | ||
| K562 | Antiproliferative activity assay | IC50 = 1.22 μM | 25778995 | ||
| MDA-MB-231 | Growth inhibition assay | GI50 = 1.26 μM | 28088086 | ||
| MDA-MB-231 | Cytotoxicity assay | GI50 = 1.26 μM | 26590508 | ||
| MDA-MB-231 | Antiproliferative activity assay | GI50 = 1.26 μM | 27017549 | ||
| SK-MEL-28 | Antiproliferative activity assay | EC50 = 1.3 μM | 18942827 | ||
| sf9 | Function assay | Kd = 1.3 μM | 28109791 | ||
| F-6-8 | Cytotoxicity assay | IC50 = 1.3 μM | 26318998 | ||
| A549 | Antiproliferative activity assay | IC50 = 1.45 μM | 26342134 | ||
| rhabdomyosarcoma | Antiviral activity assay | EC50 = 1.5 μM | 27288186 | ||
| HL-60(TB) | Growth inhibition assay | GI50 = 1.58 μM | 28088086 | ||
| RPMI8226 | Growth inhibition assay | GI50 = 1.58 μM | 28088086 | ||
| KM12 | Growth inhibition assay | GI50 = 1.58 μM | 28088086 | ||
| LOXIMVI | Growth inhibition assay | GI50 = 1.58 μM | 28088086 | ||
| HCT116 | Growth inhibition assay | GI50 = 1.58 μM | 28088086 | ||
| HOP92 | Growth inhibition assay | GI50 = 1.58 μM | 28088086 | ||
| SF539 | Growth inhibition assay | GI50 = 1.58 μM | 28088086 | ||
| UACC62 | Growth inhibition assay | GI50 = 1.58 μM | 28088086 | ||
| SF295 | Growth inhibition assay | GI50 = 1.58 μM | 28088086 | ||
| MDA-MB-435 | Growth inhibition assay | GI50 = 1.58 μM | 28088086 | ||
| SK-MEL-5 | Growth inhibition assay | GI50 = 1.58 μM | 28088086 | ||
| T47D | Growth inhibition assay | GI50 = 1.58 μM | 28088086 | ||
| RPMI8266 | Cytotoxicity assay | GI50 = 1.58 μM | 26590508 | ||
| HL-60(TB) | Cytotoxicity assay | GI50 = 1.58 μM | 26590508 | ||
| KM12 | Cytotoxicity assay | GI50 = 1.58 μM | 26590508 | ||
| HCT116 | Cytotoxicity assay | GI50 = 1.58 μM | 26590508 | ||
| SF295 | Cytotoxicity assay | GI50 = 1.58 μM | 26590508 | ||
| SF539 | Cytotoxicity assay | GI50 = 1.58 μM | 26590508 | ||
| SK-MEL-2 | Cytotoxicity assay | GI50 = 1.58 μM | 26590508 | ||
| MDA-MB-435 | Cytotoxicity assay | GI50 = 1.58 μM | 26590508 | ||
| SK-MEL-5 | Cytotoxicity assay | GI50 = 1.58 μM | 26590508 | ||
| LOXIMVI | Cytotoxicity assay | GI50 = 1.58 μM | 26590508 | ||
| T47D | Cytotoxicity assay | GI50 = 1.58 μM | 26590508 | ||
| SMMC7721 | Antiproliferative activity assay | IC50 = 1.58 μM | 26753815 | ||
| LOXIMVI | Antiproliferative activity assay | GI50 = 1.58 μM | 27017549 | ||
| UACC62 | Antiproliferative activity assay | GI50 = 1.58 μM | 27017549 | ||
| SK-MEL-5 | Antiproliferative activity assay | GI50 = 1.58 μM | 27017549 | ||
| KM12 | Antiproliferative activity assay | GI50 = 1.58 μM | 27017549 | ||
| MDA-MB-435 | Antiproliferative activity assay | GI50 = 1.58 μM | 27017549 | ||
| SF539 | Antiproliferative activity assay | GI50 = 1.58 μM | 27017549 | ||
| SF295 | Antiproliferative activity assay | GI50 = 1.58 μM | 27017549 | ||
| HCT116 | Antiproliferative activity assay | GI50 = 1.58 μM | 27017549 | ||
| HOP92 | Antiproliferative activity assay | GI50 = 1.58 μM | 27017549 | ||
| RPMI8226 | Antiproliferative activity assay | GI50 = 1.58 μM | 27017549 | ||
| HL-60(TB) | Antiproliferative activity assay | GI50 = 1.58 μM | 27017549 | ||
| T47D | Antiproliferative activity assay | GI50 = 1.58 μM | 27017549 | ||
| 7dF3 | Function assay | IC50 = 1.62 μM | 29266937 | ||
| MDA-MB-435 | Antiproliferative activity assay | IC50 = 1.67 μM | 25778995 | ||
| HL60 | Cytotoxicity assay | IC50 = 1.68 μM | 24858546 | ||
| SW579 | Antiproliferative activity human | IC50 = 1.73 μM | 28242553 | ||
| MCF7 | Antiproliferative activity assay | IC50 = 1.88 μM | 25637123 | ||
| A375P | Antiproliferative activity assay | IC50 = 1.9 μM | 24128410 | ||
| A549 | Cytotoxicity assay | IC50 = 1.92 μM | 24826815 | ||
| HCT116 | Antiproliferative activity assay | IC50 = 2 μM | 22808911 | ||
| MDA-MB-231 | Cytotoxicity assay | IC50 = 2 μM | 23454017 | ||
| NCI-H23 | Growth inhibition assay | GI50 = 2 μM | 28088086 | ||
| NCI-H522 | Growth inhibition assay | GI50 = 2 μM | 28088086 | ||
| HOP62 | Growth inhibition assay | GI50 = 2 μM | 28088086 | ||
| NCI-H226 | Growth inhibition assay | GI50 = 2 μM | 28088086 | ||
| HT-29 | Growth inhibition assay | GI50 = 2 μM | 28088086 | ||
| U251 | Growth inhibition assay | GI50 = 2 μM | 28088086 | ||
| COLO205 | Growth inhibition assay | GI50 = 2 μM | 28088086 | ||
| CCRF-CEM | Growth inhibition assay | GI50 = 2 μM | 28088086 | ||
| MALME-3M | Growth inhibition assay | GI50 = 2 μM | 28088086 | ||
| M14 | Growth inhibition assay | GI50 = 2 μM | 28088086 | ||
| UACC257 | Growth inhibition assay | GI50 = 2 μM | 28088086 | ||
| PC3 | Growth inhibition assay | GI50 = 2 μM | 28088086 | ||
| SK-MEL-2 | Growth inhibition assay | GI50 = 2 μM | 28088086 | ||
| MDA-MB-468 | Growth inhibition assay | GI50 = 2 μM | 28088086 | ||
| CCRF-CEM | Cytotoxicity assay | GI50 = 2 μM | 26590508 | ||
| NCI-H23 | Cytotoxicity assay | GI50 = 2 μM | 26590508 | ||
| NCI-H522 | Cytotoxicity assay | GI50 = 2 μM | 26590508 | ||
| COLO205 | Cytotoxicity assay | GI50 = 2 μM | 26590508 | ||
| HT-29 | Cytotoxicity assay | GI50 = 2 μM | 26590508 | ||
| SK-MEL-28 | Cytotoxicity assay | GI50 = 2 μM | 26590508 | ||
| HOP62 | Cytotoxicity assay | GI50 = 2 μM | 26590508 | ||
| UACC257 | Cytotoxicity assay | GI50 = 2 μM | 26590508 | ||
| NCI-H226 | Cytotoxicity assay | GI50 = 2 μM | 26590508 | ||
| U251 | Cytotoxicity assay | GI50 = 2 μM | 26590508 | ||
| PC3 | Cytotoxicity assay | GI50 = 2 μM | 26590508 | ||
| M14 | Cytotoxicity assay | GI50 = 2 μM | 26590508 | ||
| MDA-MB-468 | Cytotoxicity assay | GI50 = 2 μM | 26590508 | ||
| PC3 | Antiproliferative activity assay | GI50 = 2 μM | 27017549 | ||
| MALME-3M | Antiproliferative activity assay | GI50 = 2 μM | 27017549 | ||
| U251 | Antiproliferative activity assay | GI50 = 2 μM | 27017549 | ||
| UACC257 | Antiproliferative activity assay | GI50 = 2 μM | 27017549 | ||
| SK-MEL-2 | Antiproliferative activity assay | GI50 = 2 μM | 27017549 | ||
| COLO205 | Antiproliferative activity assay | GI50 = 2 μM | 27017549 | ||
| NCI-H23 | Antiproliferative activity assay | GI50 = 2 μM | 27017549 | ||
| HT-29 | Antiproliferative activity assay | GI50 = 2 μM | 27017549 | ||
| M14 | Antiproliferative activity assay | GI50 = 2 μM | 27017549 | ||
| NCI-H226 | Antiproliferative activity assay | GI50 = 2 μM | 27017549 | ||
| HOP62 | Antiproliferative activity assay | GI50 = 2 μM | 27017549 | ||
| MDA-MB-468 | Antiproliferative activity assay | GI50 = 2 μM | 27017549 | ||
| CCRF-CEM | Antiproliferative activity assay | GI50 = 2 μM | 27017549 | ||
| NCI-H522 | Antiproliferative activity assay | GI50 = 2 μM | 27017549 | ||
| A549 | Antiproliferative activity assay | IC50 = 2.02 μM | 26753815 | ||
| MDA-MB-435 | Growth inhibition assay | IC50 = 2.11 μM | 29189002 | ||
| NCI-H460 | Antiproliferative activity assay | IC50 = 2.19 μM | 30216849 | ||
| H460 | Cytotoxicity assay | IC50 = 2.19 μM | 24826815 | ||
| H460 | Cytotoxicity assay | IC50 = 2.19 μM | 25086238 | ||
| H460 | Cytotoxicity assay | IC50 = 2.19 μM | 25440879 | ||
| A375 | Toxicity assay | IC50 = 2.2 μM | 19654408 | ||
| H460 | Antiproliferative activity assay | IC50 = 2.25 μM | 26991938 | ||
| HCT116 | Antiproliferative activity assay | IC50 = 2.3 μM | 23260578 | ||
| MKN45 | Antiproliferative activity assay | IC50 = 2.32 μM | 30216849 | ||
| MKN45 | Cytotoxicity assay | IC50 = 2.32 μM | 24826815 | ||
| MKN45 | Cytotoxicity assay | IC50 = 2.32 μM | 25086238 | ||
| MKN45 | Cytotoxicity assay | IC50 = 2.32 μM | 25440879 | ||
| U937 | Antiproliferative activity assay | GI50 = 2.34 μM | 29459144 | ||
| A375 | Antiproliferative activity assay | IC50 = 2.4 μM | 22808911 | ||
| HeLa | Growth inhibition assay | IC50 = 2.44 μM | 29102175 | ||
| MDA-MB-231 | Cytotoxicity assay | IC50 = 2.5 μM | 22414612 | ||
| MCF7 | Antiproliferative activity assay | IC50 = 2.51 μM | 30216849 | ||
| UO31 | Antiproliferative activity assay | IC50 = 2.51 μM | 30216849 | ||
| NCI-H322M | Growth inhibition assay | GI50 = 2.51 μM | 28088086 | ||
| NCI-H460 | Growth inhibition assay | GI50 = 2.51 μM | 28088086 | ||
| KM12 | Growth inhibition assay | TGI = 2.51 μM | 28088086 | ||
| LOXIMVI | Growth inhibition assay | TGI = 2.51 μM | 28088086 | ||
| EKVX | Growth inhibition assay | GI50 = 2.51 μM | 28088086 | ||
| HCT116 | Growth inhibition assay | TGI = 2.51 μM | 28088086 | ||
| SW620 | Growth inhibition assay | GI50 = 2.51 μM | 28088086 | ||
| SF268 | Growth inhibition assay | GI50 = 2.51 μM | 28088086 | ||
| SF539 | Growth inhibition assay | TGI = 2.51 μM | 28088086 | ||
| A498 | Growth inhibition assay | GI50 = 2.51 μM | 28088086 | ||
| UACC62 | Growth inhibition assay | TGI = 2.51 μM | 28088086 | ||
| IGROV1 | Growth inhibition assay | GI50 = 2.51 μM | 28088086 | ||
| HCT15 | Growth inhibition assay | GI50 = 2.51 μM | 28088086 | ||
| SK-MEL-28 | Growth inhibition assay | GI50 = 2.51 μM | 28088086 | ||
| SK-MEL-28 | Growth inhibition assay | TGI = 2.51 μM | 28088086 | ||
| ACHN | Growth inhibition assay | GI50 = 2.51 μM | 28088086 | ||
| SK-MEL-5 | Growth inhibition assay | TGI = 2.51 μM | 28088086 | ||
| NCI/ADR-RES | Growth inhibition assay | GI50 = 2.51 μM | 28088086 | ||
| MCF7 | Growth inhibition assay | GI50 = 2.51 μM | 28088086 | ||
| SN12C | Growth inhibition assay | GI50 = 2.51 μM | 28088086 | ||
| Hs 578T | Growth inhibition assay | GI50 = 2.51 μM | 28088086 | ||
| SKOV3 | Growth inhibition assay | GI50 = 2.51 μM | 28088086 | ||
| UO31 | Growth inhibition assay | GI50 = 2.51 μM | 28088086 | ||
| NCI-H460 | Cytotoxicity assay | GI50 = 2.51 μM | 26590508 | ||
| HCT15 | Cytotoxicity assay | GI50 = 2.51 μM | 26590508 | ||
| SW620 | Cytotoxicity assay | GI50 = 2.51 μM | 26590508 | ||
| SF268 | Cytotoxicity assay | GI50 = 2.51 μM | 26590508 | ||
| IGROV1 | Cytotoxicity assay | GI50 = 2.51 μM | 26590508 | ||
| OVCAR8 | Cytotoxicity assay | GI50 = 2.51 μM | 26590508 | ||
| NCI/ADR-RES | Cytotoxicity assay | GI50 = 2.51 μM | 26590508 | ||
| SKOV3 | Cytotoxicity assay | GI50 = 2.51 μM | 26590508 | ||
| SN12C | Cytotoxicity assay | GI50 = 2.51 μM | 26590508 | ||
| NCI-H322M | Cytotoxicity assay | GI50 = 2.51 μM | 26590508 | ||
| MCF7 | Cytotoxicity assay | GI50 = 2.51 μM | 26590508 | ||
| UO31 | Cytotoxicity assay | GI50 = 2.51 μM | 26590508 | ||
| Hs578T | Cytotoxicity assay | GI50 = 2.51 μM | 26590508 | ||
| A498 | Cytotoxicity assay | GI50 = 2.51 μM | 26590508 | ||
| A498 | Antiproliferative activity assay | GI50 = 2.51 μM | 27017549 | ||
| SKOV3 | Antiproliferative activity assay | GI50 = 2.51 μM | 27017549 | ||
| UO31 | Antiproliferative activity assay | GI50 = 2.51 μM | 27017549 | ||
| NCI-ADR-RES | Antiproliferative activity assay | GI50 = 2.51 μM | 27017549 | ||
| SN12C | Antiproliferative activity assay | GI50 = 2.51 μM | 27017549 | ||
| IGROV1 | Antiproliferative activity assay | GI50 = 2.51 μM | 27017549 | ||
| ACHN | Antiproliferative activity assay | GI50 = 2.51 μM | 27017549 | ||
| OVCAR8 | Antiproliferative activity assay | GI50 = 2.51 μM | 27017549 | ||
| SK-MEL-28 | Antiproliferative activity assay | GI50 = 2.51 μM | 27017549 | ||
| NCI-H322M | Antiproliferative activity assay | GI50 = 2.51 μM | 27017549 | ||
| SF268 | Antiproliferative activity assay | GI50 = 2.51 μM | 27017549 | ||
| HCT15 | Antiproliferative activity assay | GI50 = 2.51 μM | 27017549 | ||
| NCI-H460 | Antiproliferative activity assay | GI50 = 2.51 μM | 27017549 | ||
| SW620 | Antiproliferative activity assay | GI50 = 2.51 μM | 27017549 | ||
| EKVX | Antiproliferative activity assay | GI50 = 2.51 μM | 27017549 | ||
| MCF7 | Antiproliferative activity assay | GI50 = 2.51 μM | 27017549 | ||
| Hs 578T | Antiproliferative activity assay | GI50 = 2.51 μM | 27017549 | ||
| HT-29 | Antiproliferative activity assay | IC50 = 2.58 μM | 29886324 | ||
| A375P | Antiproliferative activity assay | GI50 = 2.58 μM | 26810260 | ||
| TPC1 | Antiproliferative activity assay | EC50 = 2.6 μM | 22559926 | ||
| Hep3B | Growth inhibition assay | IC50 = 2.63 μM | 29102175 | ||
| A375P | Antiproliferative activity assay | IC50 = 2.7 μM | 22460030 | ||
| U937 | Antiproliferative activity assay | GI50 = 2.74 μM | 26318067 | ||
| U937 | Cytotoxicity assay | GI50 = 2.74 μM | 24878193 | ||
| MCF7 | Growth inhibition assay | IC50 = 2.78 μM | 29102175 | ||
| MDA-MB-231 | Antiproliferative activity assay | IC50 = 2.8 μM | 29202403 | ||
| HepG2 | Antiproliferative activity assay | IC50 = 2.84 μM | 28242553 | ||
| U937 | Growth inhibition assay | GI50 = 2.85 μM | 22014755 | ||
| A549 | Cytotoxicity assay | IC50 = 2.92 μM | 28927801 | ||
| A549 | Cytotoxicity assay | IC50 = 2.92 μM | 27777009 | ||
| ZR75-30 | Growth inhibition assay | IC50 = 2.96 μM | 29102175 | ||
| PC3 | Cytotoxicity assay | IC50 = 3.03 μM | 28340913 | ||
| MDA-MB-231 | Antiproliferative activity assay | IC50 = 3.08 μM | 26991938 | ||
| A549 | Growth inhibition assay | IC50 = 3.1 μM | 29102175 | ||
| K562 | Growth inhibition assay | GI50 = 3.16 μM | 28088086 | ||
| MOLT4 | Growth inhibition assay | GI50 = 3.16 μM | 28088086 | ||
| A549/ATCC | Growth inhibition assay | GI50 = 3.16 μM | 28088086 | ||
| RPMI8226 | Growth inhibition assay | TGI = 3.16 μM | 28088086 | ||
| SR | Growth inhibition assay | GI50 = 3.16 μM | 28088086 | ||
| HOP62 | Growth inhibition assay | TGI = 3.16 μM | 28088086 | ||
| NCI-H226 | Growth inhibition assay | TGI = 3.16 μM | 28088086 | ||
| U251 | Growth inhibition assay | TGI = 3.16 μM | 28088086 | ||
| COLO205 | Growth inhibition assay | TGI = 3.16 μM | 28088086 | ||
| M14 | Growth inhibition assay | TGI = 3.16 μM | 28088086 | ||
| HCC2998 | Growth inhibition assay | GI50 = 3.16 μM | 28088086 | ||
| SNB75 | Growth inhibition assay | GI50 = 3.16 μM | 28088086 | ||
| 786-0 | Growth inhibition assay | GI50 = 3.16 μM | 28088086 | ||
| A498 | Growth inhibition assay | TGI = 3.16 μM | 28088086 | ||
| HCT15 | Growth inhibition assay | TGI = 3.16 μM | 28088086 | ||
| OVCAR3 | Growth inhibition assay | GI50 = 3.16 μM | 28088086 | ||
| OVCAR4 | Growth inhibition assay | GI50 = 3.16 μM | 28088086 | ||
| Caki1 | Growth inhibition assay | GI50 = 3.16 μM | 28088086 | ||
| SNB19 | Growth inhibition assay | GI50 = 3.16 μM | 28088086 | ||
| OVCAR5 | Growth inhibition assay | GI50 = 3.16 μM | 28088086 | ||
| DU145 | Growth inhibition assay | GI50 = 3.16 μM | 28088086 | ||
| OVCAR8 | Growth inhibition assay | GI50 = 3.16 μM | 28088086 | ||
| SN12C | Growth inhibition assay | TGI = 3.16 μM | 28088086 | ||
| MDA-MB-231 | Growth inhibition assay | TGI = 3.16 μM | 28088086 | ||
| RXF393 | Growth inhibition assay | GI50 = 3.16 μM | 28088086 | ||
| BT549 | Growth inhibition assay | GI50 = 3.16 μM | 28088086 | ||
| K562 | Cytotoxicity assay | GI50 = 3.16 μM | 26590508 | ||
| MOLT4 | Cytotoxicity assay | GI50 = 3.16 μM | 26590508 | ||
| SNB19 | Cytotoxicity assay | GI50 = 3.16 μM | 26590508 | ||
| SR | Cytotoxicity assay | GI50 = 3.16 μM | 26590508 | ||
| A549 | Cytotoxicity assay | GI50 = 3.16 μM | 26590508 | ||
| OVCAR3 | Cytotoxicity assay | GI50 = 3.16 μM | 26590508 | ||
| OVCAR4 | Cytotoxicity assay | GI50 = 3.16 μM | 26590508 | ||
| SNB75 | Cytotoxicity assay | GI50 = 3.16 μM | 26590508 | ||
| OVCAR5 | Cytotoxicity assay | GI50 = 3.16 μM | 26590508 | ||
| DU145 | Cytotoxicity assay | GI50 = 3.16 μM | 26590508 | ||
| 786-0 | Cytotoxicity assay | GI50 = 3.16 μM | 26590508 | ||
| BT549 | Cytotoxicity assay | GI50 = 3.16 μM | 26590508 | ||
| ACHN | Cytotoxicity assay | GI50 = 3.16 μM | 26590508 | ||
| Caki1 | Cytotoxicity assay | GI50 = 3.16 μM | 26590508 | ||
| 786-0 | Antiproliferative activity assay | GI50 = 3.16 μM | 27017549 | ||
| LOXIMVI | Antiproliferative activity assay | TGI = 3.16 μM | 27017549 | ||
| Caki1 | Antiproliferative activity assay | GI50 = 3.16 μM | 27017549 | ||
| OVCAR5 | Antiproliferative activity assay | GI50 = 3.16 μM | 27017549 | ||
| RXF393 | Antiproliferative activity assay | GI50 = 3.16 μM | 27017549 | ||
| SNB75 | Antiproliferative activity assay | GI50 = 3.16 μM | 27017549 | ||
| OVCAR3 | Antiproliferative activity assay | GI50 = 3.16 μM | 27017549 | ||
| SNB19 | Antiproliferative activity assay | GI50 = 3.16 μM | 27017549 | ||
| SK-MEL-5 | Antiproliferative activity assay | TGI = 3.16 μM | 27017549 | ||
| OVCAR4 | Antiproliferative activity assay | GI50 = 3.16 μM | 27017549 | ||
| SK-MEL-2 | Antiproliferative activity assay | TGI = 3.16 μM | 27017549 | ||
| BT549 | Antiproliferative activity assay | GI50 = 3.16 μM | 27017549 | ||
| MOLT4 | Antiproliferative activity assay | GI50 = 3.16 μM | 27017549 | ||
| K562 | Antiproliferative activity assay | GI50 = 3.16 μM | 27017549 | ||
| HCC2998 | Antiproliferative activity assay | GI50 = 3.16 μM | 27017549 | ||
| HCC2998 | Antiproliferative activity assay | TGI = 3.16 μM | 27017549 | ||
| A549/ATCC | Antiproliferative activity assay | GI50 = 3.16 μM | 27017549 | ||
| DU145 | Antiproliferative activity assay | GI50 = 3.16 μM | 27017549 | ||
| SR | Antiproliferative activity assay | GI50 = 3.16 μM | 27017549 | ||
| MCF7 | Cytotoxicity assay | IC50 = 3.18 μM | 28927801 | ||
| PC3 | Cytotoxicity assay | IC50 = 3.18 μM | 28927801 | ||
| MCF7 | Cytotoxicity assay | IC50 = 3.18 μM | 27777009 | ||
| A549 | Antiproliferative activity assay | IC50 = 3.19 μM | 25778995 | ||
| PC3 | Cytotoxicity assay | IC50 = 3.24 μM | 27777009 | ||
| HeLa | Function assay | IC50 = 3.3 μM | 15225706 | ||
| HeLa | Function assay | IC50 = 3.3 μM | 15225706 | ||
| LoVo | Cytotoxicity assay | IC50 = 3.3 μM | 23454017 | ||
| A375 | Antiproliferative activity assay | IC50 = 3.36 μM | 29602674 | ||
| HT-29 | Antiproliferative activity assay | IC50 = 3.37 μM | 26991938 | ||
| FLT3 gene-deficient U937 | Antiproliferative activity assay | GI50 = 3.4 μM | 21708468 | ||
| FLT3 negative U937 | Cytotoxicity assay | GI50 = 3.4 μM | 23618709 | ||
| U937 | Cytotoxicity assay | GI50 = 3.4 μM | 26081023 | ||
| A375P | Antiproliferative activity assay | GI50 = 3.4 μM | 29459144 | ||
| ACHN | Cytotoxicity assay | IC50 = 3.4 μM | 29517908 | ||
| ACHN | Cytotoxicity assay | IC50 = 3.4 μM | 29297688 | ||
| HepG2 | Function assay | IC50 = 3.4 μM | 26071861 | ||
| A375P | Cytotoxicity assay | GI50 = 3.4 μM | 24878193 | ||
| HepG2 | Cytotoxicity assay | IC50 = 3.44 μM | 28927801 | ||
| HepG2 | Cytotoxicity assay | IC50 = 3.44 μM | 27777009 | ||
| ACHN | Cytotoxicity assay | IC50 = 3.5 μM | 29786436 | ||
| HepG2 | Antiproliferative activity assay | IC50 = 3.5 μM | 25462265 | ||
| HT-29 | Cytotoxicity assay | IC50 = 3.61 μM | 24826815 | ||
| HT-29 | Antiproliferative activity assay | IC50 = 3.61 μM | 30216849 | ||
| HT-29 | Cytotoxicity assay | IC50 = 3.61 μM | 25086238 | ||
| HT-29 | Cytotoxicity assay | IC50 = 3.61 μM | 25440879 | ||
| MCF7 | Anticancer activity assay | IC50 = 3.64 μM | 29549841 | ||
| Sf9 | Function assay | IC50 = 3.8 μM | 21708468 | ||
| Sf9 | Function assay | IC50 = 3.8 μM | 23618709 | ||
| Sf9 | Function assay | IC50 = 3.8 μM | 26081023 | ||
| A2058 | Cytotoxicity assay | IC50 = 3.8 μM | 22708987 | ||
| NCI-H460 | Cytotoxicity assay | IC50 = 3.9 μM | 23454017 | ||
| SK-MEL-30 | Cytotoxicity assay | IC50 = 3.9 μM | 29461827 | ||
| HT-29 | Anticancer activity assay | IC50 = 3.97 μM | 29549841 | ||
| SF268 | Growth inhibition assay | TGI = 3.98 μM | 28088086 | ||
| SNB75 | Growth inhibition assay | TGI = 3.98 μM | 28088086 | ||
| OVCAR3 | Growth inhibition assay | TGI = 3.98 μM | 28088086 | ||
| SNB19 | Growth inhibition assay | TGI = 3.98 μM | 28088086 | ||
| Hs 578T | Growth inhibition assay | TGI = 3.98 μM | 28088086 | ||
| TK10 | Growth inhibition assay | GI50 = 3.98 μM | 28088086 | ||
| BT549 | Growth inhibition assay | TGI = 3.98 μM | 28088086 | ||
| Caki1 | Antiproliferative activity assay | TGI = 3.98 μM | 27017549 | ||
| TK10 | Antiproliferative activity assay | GI50 = 3.98 μM | 27017549 | ||
| MDA-MB-435 | Antiproliferative activity assay | TGI = 3.98 μM | 27017549 | ||
| HCT116 | Antiproliferative activity assay | TGI = 3.98 μM | 27017549 | ||
| MDA-MB-231 | Antiproliferative activity assay | TGI = 3.98 μM | 27017549 | ||
| HepG2 | Cytotoxicity assay | IC50 = 4 μM | 23454017 | ||
| HT-29 | Cytotoxicity assay | IC50 = 4 μM | 23454017 | ||
| A549 | Cytotoxicity assay | IC50 = 4 μM | 23454017 | ||
| HuH7 | Cytotoxicity assay | GI50 = 4 μM | 23726028 | ||
| PC3 | Antiproliferative activity assay | IC50 = 4.13 μM | 25778995 | ||
| HeLa | Cytotoxicity assay | IC50 = 4.163 μM | 23362959 | ||
| MDA-MB-436 | Antiproliferative activity assay | IC50 = 4.2 μM | 29202403 | ||
| MCF7 | Cytotoxicity assay | IC50 = 4.21 μM | 27043268 | ||
| MCF7 | Cytotoxicity assay | IC50 = 4.21 μM | 28340913 | ||
| LOXIMVI | Antiproliferative activity assay | IC50 = 4.25 μM | 25778995 | ||
| OVCAR4 | Cytotoxicity assay | TGI = 4.31 μM | 26590508 | ||
| WM1361 | Antiproliferative activity assay | GI50 = 4.345 μM | 20148563 | ||
| WM266.4 | Growth inhibition assay | GI50 = 4.5 μM | 20199087 | ||
| HepG2 | Cytotoxicity assay | GI50 = 4.5 μM | 23726028 | ||
| A549 | Antiproliferative activity assay | IC50 = 4.5 μM | 23260578 | ||
| LS174T | Function assay | IC50 = 4.52 μM | 29266937 | ||
| 8505C | Antiproliferative activity assay | IC50 = 4.7 μM | 29032031 | ||
| OVCAR8 | Cytotoxicity assay | TGI = 4.73 μM | 26590508 | ||
| MDA-MB-231 | Cytotoxicity assay | IC50 = 4.77 μM | 20435479 | ||
| B16-F1 | Antiproliferative activity assay | IC50 = 4.9 μM | 18477505 | ||
| WM164 | Antiproliferative activity assay | IC50 = 4.9 μM | 17561392 | ||
| B16-F1 | Antiproliferative activity assay | IC50 = 4.9 μM | 20056548 | ||
| MDA-MB-468 | Cytotoxicity assay | IC50 = 4.9 μM | 26159483 | ||
| 786-O | Cytotoxicity assay | IC50 = 4.9 μM | 29517908 | ||
| 786-O | Cytotoxicity assay | IC50 = 4.9 μM | 29297688 | ||
| WM266.4 | Antiproliferative activity assay | GI50 = 4.933 μM | 20148563 | ||
| WM266.4 | Growth inhibition assay | IC50 = 5 μM | 19323560 | ||
| WM164 | Antiproliferative activity assay | IC50 = 5 μM | 20056548 | ||
| HepG2 | Antiproliferative activity assay | IC50 = 5 μM | 23260578 | ||
| HepG2 | Antiproliferative activity assay | IC50 = 5 μM | 23932071 | ||
| NCI-H23 | Growth inhibition assay | TGI = 5.01 μM | 28088086 | ||
| NCI-H322M | Growth inhibition assay | TGI = 5.01 μM | 28088086 | ||
| NCI-H460 | Growth inhibition assay | TGI = 5.01 μM | 28088086 | ||
| NCI-H522 | Growth inhibition assay | TGI = 5.01 μM | 28088086 | ||
| HT-29 | Growth inhibition assay | TGI = 5.01 μM | 28088086 | ||
| CCRF-CEM | Growth inhibition assay | TGI = 5.01 μM | 28088086 | ||
| SW620 | Growth inhibition assay | TGI = 5.01 μM | 28088086 | ||
| HOP92 | Growth inhibition assay | TGI = 5.01 μM | 28088086 | ||
| 786-0 | Growth inhibition assay | TGI = 5.01 μM | 28088086 | ||
| MDA-MB-435 | Growth inhibition assay | TGI = 5.01 μM | 28088086 | ||
| PC3 | Growth inhibition assay | TGI = 5.01 μM | 28088086 | ||
| UACC257 | Growth inhibition assay | TGI = 5.01 μM | 28088086 | ||
| SKOV3 | Growth inhibition assay | TGI = 5.01 μM | 28088086 | ||
| RXF393 | Growth inhibition assay | TGI = 5.01 μM | 28088086 | ||
| T47D | Growth inhibition assay | TGI = 5.01 μM | 28088086 | ||
| MDA-MB-468 | Growth inhibition assay | TGI = 5.01 μM | 28088086 | ||
| UACC62 | Antiproliferative activity assay | TGI = 5.01 μM | 27017549 | ||
| UACC257 | Antiproliferative activity assay | TGI = 5.01 μM | 27017549 | ||
| SK-MEL-5 | Antiproliferative activity assay | LC50 = 5.01 μM | 27017549 | ||
| M14 | Antiproliferative activity assay | TGI = 5.01 μM | 27017549 | ||
| NCI-H23 | Antiproliferative activity assay | TGI = 5.01 μM | 27017549 | ||
| SF295 | Antiproliferative activity assay | TGI = 5.01 μM | 27017549 | ||
| HOP92 | Antiproliferative activity assay | TGI = 5.01 μM | 27017549 | ||
| BT549 | Antiproliferative activity assay | TGI = 5.01 μM | 27017549 | ||
| MDA-MB-231 | Cytotoxicity assay | IC50 = 5.1 μM | 26159483 | ||
| DU145 | Cytotoxicity assay | IC50 = 5.1 μM | 22708987 | ||
| A549 | Cytotoxicity assay | IC50 = 5.21 μM | 20181414 | ||
| HeLa | Cytotoxicity assay | IC50 = 5.23 μM | 26342867 | ||
| 786-O | Cytotoxicity assay | IC50 = 5.3 μM | 29786436 | ||
| HCT116 | Antiproliferative activity assay | GI50 = 5.4 μM | 15225706 | ||
| A375 | Antiproliferative activity assay | IC50 = 5.4 μM | 18477505 | ||
| SK-MEL-188 | Antiproliferative activity assay | IC50 = 5.4 μM | 17561392 | ||
| A375 | Antiproliferative activity assay | IC50 = 5.4 μM | 20056548 | ||
| MCF7 | Cytotoxicity assay | IC50 = 5.5 μM | 26159483 | ||
| A375 | Antiproliferative activity assay | IC50 = 5.58 μM | 19464887 | ||
| A375P | Cytotoxicity assay | IC50 = 5.58 μM | 20797858 | ||
| A375P | Antiproliferative activity assay | GI50 = 5.58 μM | 21353571 | ||
| A375P | Growth inhibition assay | GI50 = 5.58 μM | 22014755 | ||
| A375P | Growth inhibition assay | GI50 = 5.58 μM | 20149658 | ||
| A375P | Antiproliferative activity assay | GI50 = 5.58 μM | 20466542 | ||
| A375P | Antiproliferative activity assay | IC50 = 5.6 μM | 19857963 | ||
| A375P | Antiproliferative activity assay | IC50 = 5.6 μM | 21592628 | ||
| A375P | Antiproliferative activity assay | IC50 = 5.6 μM | 22033063 | ||
| A375P | Cytotoxicity assay | IC50 = 5.6 μM | 19897366 | ||
| A375P | Antiproliferative activity assay | IC50 = 5.6 μM | 22647720 | ||
| HCT116 | Cytotoxicity assay | IC50 = 5.65 μM | 24215818 | ||
| SH-SY5Y | Antiproliferative activity assay | IC50 = 5.73 μM | 24315192 | ||
| HepG2 | Cytotoxicity assay | IC50 = 5.74 μM | 25461318 | ||
| Bel7402 | Cytotoxicity assay | IC50 = 5.8 μM | 23454017 | ||
| HT-29 | Cytotoxicity assay | IC50 = 5.9 μM | 28865276 | ||
| melanoma | Growth inhibition assay | IC50 = 6.1 μM | 16392826 | ||
| K562 | Antiproliferative activity assay | GI50 = 6.2 μM | 21708468 | ||
| HepG2 | Cytotoxicity assay | IC50 = 6.2 μM | 28865276 | ||
| A549 | Anticancer activity assay | IC50 = 6.21 μM | 29549841 | ||
| SMMC7721 | Antiproliferative activity assay | IC50 = 6.23 μM | 22721924 | ||
| HepG2 | Antiproliferative activity assay | IC50 = 6.3 μM | 23932071 | ||
| SF295 | Growth inhibition assay | TGI = 6.31 μM | 28088086 | ||
| SK-MEL-2 | Growth inhibition assay | TGI = 6.31 μM | 28088086 | ||
| OVCAR5 | Growth inhibition assay | TGI = 6.31 μM | 28088086 | ||
| UO31 | Growth inhibition assay | TGI = 6.31 μM | 28088086 | ||
| A498 | Antiproliferative activity assay | TGI = 6.31 μM | 27017549 | ||
| MALME-3M | Antiproliferative activity assay | TGI = 6.31 μM | 27017549 | ||
| U251 | Antiproliferative activity assay | TGI = 6.31 μM | 27017549 | ||
| SK-MEL-2 | Antiproliferative activity assay | LC50 = 6.31 μM | 27017549 | ||
| RPMI8226 | Antiproliferative activity assay | TGI = 6.31 μM | 27017549 | ||
| HCC2998 | Antiproliferative activity assay | LC50 = 6.31 μM | 27017549 | ||
| NCI-H522 | Antiproliferative activity assay | TGI = 6.31 μM | 27017549 | ||
| HUVEC | Antiproliferative activity assay | IC50 = 6.42 μM | 23644219 | ||
| A549 | Cytotoxicity assay | IC50 = 6.53 μM | 27043268 | ||
| A549 | Cytotoxicity assay | IC50 = 6.53 μM | 28340913 | ||
| LoVo | Antiproliferative activity assay | IC50 = 6.56 μM | 24315192 | ||
| A375 | Function assay | IC50 = 6.599 μM | 21807507 | ||
| HCT116 | Function assay | GI50 = 6.6 μM | 15225706 | ||
| HepG2 | Cytotoxicity assay | IC50 = 6.7 μM | 26159483 | ||
| BCPAP | Antiproliferative activity assay | IC50 = 6.7 μM | 29032031 | ||
| A549 | Cytotoxicity assay | IC50 = 6.7 μM | 28865276 | ||
| PC3 | Cytotoxicity assay | IC50 = 6.8 μM | 23021967 | ||
| SGC7901 | Cytotoxicity assay | IC50 = 6.9 μM | 23454017 | ||
| OS-RC2 | Cytotoxicity assay | IC50 = 7 μM | 29517908 | ||
| OS-RC2 | Cytotoxicity assay | IC50 = 7 μM | 29297688 | ||
| COLO205 | Antiproliferative activity human | IC50 = 7.04 μM | 28242553 | ||
| rhabdomyosarcoma | Cytotoxicity activity against human | CC50 = 7.05 μM | 27288186 | ||
| HuH7 | Cytotoxicity assay | IC50 = 7.1 μM | 26159483 | ||
| MDA-MB-231 | Antiproliferative activity assay | IC50 = 7.18 μM | 22721924 | ||
| FLT3-negative K562 | Cytotoxicity assay | GI50 = 7.3 μM | 23618709 | ||
| K562 | Function assay | GI50 = 7.3 μM | 26081023 | ||
| K562 | Function assay | GI50 = 7.3 μM | 26081023 | ||
| MCF7 | Antiproliferative activity assay | IC50 = 7.33 μM | 23644219 | ||
| A549 | Cytotoxicity assay | TGI = 7.36 μM | 26590508 | ||
| WI38 | Cytotoxicity assay | IC50 = 7.54 μM | 24826815 | ||
| A375 | Cytotoxicity assay | IC50 = 7.56 μM | 24215818 | ||
| MDA-MB-231 | Cytotoxicity assay | IC50 = 7.62 μM | 20181414 | ||
| 786-0 | Cytotoxicity assay | TGI = 7.62 μM | 26590508 | ||
| HS27 | Antiproliferative activity assay | IC50 = 7.8 μM | 19857963 | ||
| HS27 | Cytotoxicity assay | IC50 = 7.8 μM | 19897366 | ||
| HCT116 | Cytotoxicity assay | IC50 = 7.8 μM | 22483592 | ||
| HS27 | Antiproliferative activity assay | IC50 = 7.85 μM | 19464887 | ||
| HS27 | Antiproliferative activity assay | GI50 = 7.85 μM | 21353571 | ||
| A549/ATCC | Growth inhibition assay | TGI = 7.94 μM | 28088086 | ||
| DU145 | Growth inhibition assay | TGI = 7.94 μM | 28088086 | ||
| NCI/ADR-RES | Growth inhibition assay | TGI = 7.94 μM | 28088086 | ||
| MCF7 | Growth inhibition assay | TGI = 7.94 μM | 28088086 | ||
| PC3 | Antiproliferative activity assay | TGI = 7.94 μM | 27017549 | ||
| UO31 | Antiproliferative activity assay | TGI = 7.94 μM | 27017549 | ||
| NCI-ADR-RES | Antiproliferative activity assay | TGI = 7.94 μM | 27017549 | ||
| LOXIMVI | Antiproliferative activity assay | LC50 = 7.94 μM | 27017549 | ||
| SK-MEL-28 | Antiproliferative activity assay | TGI = 7.94 μM | 27017549 | ||
| KM12 | Antiproliferative activity assay | TGI = 7.94 μM | 27017549 | ||
| COLO205 | Antiproliferative activity assay | TGI = 7.94 μM | 27017549 | ||
| HT-29 | Antiproliferative activity assay | TGI = 7.94 μM | 27017549 | ||
| NCI-H226 | Antiproliferative activity assay | TGI = 7.94 μM | 27017549 | ||
| HOP62 | Antiproliferative activity assay | TGI = 7.94 μM | 27017549 | ||
| T47D | Antiproliferative activity assay | TGI = 7.94 μM | 27017549 | ||
| MDA-MB-468 | Antiproliferative activity assay | TGI = 7.94 μM | 27017549 | ||
| EKVX | Antiproliferative activity assay | TGI = 7.94 μM | 27017549 | ||
| MCF7 | Antiproliferative activity assay | TGI = 7.94 μM | 27017549 | ||
| Hs 578T | Antiproliferative activity assay | TGI = 7.94 μM | 27017549 | ||
| MGC803 | Antiproliferative activity assay | IC50 = 7.99 μM | 26560049 | ||
| HCT116 | Antiproliferative activity assay | IC50 = 8.08 μM | 24440479 | ||
| PC3 | Cytotoxicity assay | IC50 = 8.08 μM | 27043268 | ||
| HCT116 | Antiproliferative activity assay | IC50 = 8.08 μM | 23644219 | ||
| WM266.4 | Antiproliferative activity assay | GI50 = 8.1 μM | 23025996 | ||
| WM266.4 | Antiproliferative activity assay | GI50 = 8.12 μM | 22583669 | ||
| Ketr3 | Antiproliferative activity assay | IC50 = 8.27 μM | 30216849 | ||
| A375 | Antiproliferative activity assay | IC50 = 8.27 μM | 30216849 | ||
| HepG2 | Antiproliferative activity assay | IC50 = 8.27 μM | 30216849 | ||
| MX1 | Antiproliferative activity assay | IC50 = 8.27 μM | 30216849 | ||
| MX1 | Cytotoxicity assay | IC50 = 8.27 μM | 24675135 | ||
| WM266.4 | Growth inhibition assay | GI50 = 8.3 μM | 22361686 | ||
| PLC/PRF/5 | Cytotoxicity assay | IC50 = 8.3 μM | 21531053 | ||
| A375 | Antiproliferative activity assay | IC50 = 8.33 μM | 29886324 | ||
| HCT116 | Cytotoxicity assay | IC50 = 8.41 μM | 29631788 | ||
| WI38 | Cytotoxicity assay | IC50 = 8.42 μM | 26991938 | ||
| HepG2 | Cytotoxicity assay | IC50 = 8.42 μM | 27162123 | ||
| HepG2 | Cytotoxicity assay | IC50 = 8.42 μM | 23871909 | ||
| HT-29 | Antiproliferative activity assay | IC50 = 8.44 μM | 29602674 | ||
| HepG2 | Cytotoxicity assay | IC50 = 8.67 μM | 24675135 | ||
| MCF7 | Cytotoxicity assay | IC50 = 8.83 μM | 20435479 | ||
| MCF7 | Cytotoxicity assay | IC50 = 9.12 μM | 24300920 | ||
| HepG2 | Antiproliferative activity assay | IC50 = 9.14 μM | 26560049 | ||
| A375 | Cytotoxicity assay | IC50 = 9.17 μM | 24675135 | ||
| MGC803 | Antiproliferative activity assay | IC50 = 9.2 μM | 29032031 | ||
| SKOV3 | Cytotoxicity assay | IC50 = 9.25 μM | 24300920 | ||
| RS4:11 | Function assay | GI50 = 9.3 μM | 26081023 | ||
| RS4:11 | Cytotoxicity assay | GI50 = 9.3 μM | 23618709 | ||
| MIAPaCa2 | Antiproliferative activity assay | IC50 = 9.32 μM | 29032031 | ||
| PANC1 | Growth inhibition assay | IC50 = 9.32 μM | 29102175 | ||
| MHCC97H | Growth inhibition assay | IC50 = 9.32 μM | 29102175 | ||
| RS4:11 | Antiproliferative activity assay | GI50 = 9.4 μM | 21708468 | ||
| Hep3B | Cytotoxicity assay | IC50 = 9.4 μM | 28865276 | ||
| LoVo | Growth inhibition assay | IC50 = 9.47 μM | 29102175 | ||
| SMMC7721 | Antiproliferative activity assay | IC50 = 9.96 μM | 25637123 | ||
| MCF7 | Cytotoxicity assay | TGI = 9.97 μM | 26590508 | ||
| IGROV1 | Growth inhibition assay | TGI = 10 μM | 28088086 | ||
| ACHN | Growth inhibition assay | TGI = 10 μM | 28088086 | ||
| TK10 | Growth inhibition assay | TGI = 10 μM | 28088086 | ||
| 786-0 | Antiproliferative activity assay | TGI = 10 μM | 27017549 | ||
| SN12C | Antiproliferative activity assay | TGI = 10 μM | 27017549 | ||
| Caki1 | Antiproliferative activity assay | LC50 = 10 μM | 27017549 | ||
| TK10 | Antiproliferative activity assay | TGI = 10 μM | 27017549 | ||
| RXF393 | Antiproliferative activity assay | TGI = 10 μM | 27017549 | ||
| OVCAR8 | Antiproliferative activity assay | TGI = 10 μM | 27017549 | ||
| SNB75 | Antiproliferative activity assay | TGI = 10 μM | 27017549 | ||
| NCI-H322M | Antiproliferative activity assay | TGI = 10 μM | 27017549 | ||
| MDA-MB-435 | Antiproliferative activity assay | LC50 = 10 μM | 27017549 | ||
| SF539 | Antiproliferative activity assay | TGI = 10 μM | 27017549 | ||
| SF268 | Antiproliferative activity assay | TGI = 10 μM | 27017549 | ||
| NCI-H460 | Antiproliferative activity assay | TGI = 10 μM | 27017549 | ||
| DU145 | Antiproliferative activity assay | TGI = 10 μM | 27017549 | ||
| K1 | Antiproliferative activity assay | IC50 = 10.2 μM | 29032031 | ||
| Bel7402 | Cytotoxicity assay | IC50 = 10.26 μM | 21504204 | ||
| Bel7402 | Growth inhibition assay | IC50 = 10.31 μM | 29102175 | ||
| OVCAR3 | Cytotoxicity assay | TGI = 10.32 μM | 26590508 | ||
| SGC7901 | Cytotoxicity assay | IC50 = 10.37 μM | 20435479 | ||
| SNB75 | Cytotoxicity assay | TGI = 10.42 μM | 26590508 | ||
| A431 | Antiproliferative activity assay | IC50 = 10.46 μM | 29032031 | ||
| SMMC7721 | Antiproliferative activity assay | IC50 = 10.61 μM | 29032031 | ||
| GES-1 | Cytotoxicity assay | IC50 = 10.68 μM | 27162123 | ||
| H460 | Cytotoxicity assay | IC50 = 10.8 μM | 24300920 | ||
| BGC823 | Antiproliferative activity assay | IC50 = 10.91 μM | 22721924 | ||
| Hep3B | Cytotoxicity assay | IC50 = 11.2 μM | 28342400 | ||
| MCF7 | Cytotoxicity assay | IC50 = 11.34 μM | 27162123 | ||
| MCF7 | Cytotoxicity assay | IC50 = 11.34 μM | 23871909 | ||
| SK-MEL-2 | Antiproliferative activity human | IC50 = 11.35 μM | 28242553 | ||
| HepG2 | Cytotoxicity assay | IC50 = 11.49 μM | 21504204 | ||
| A375P | Antiproliferative activity assay | IC50 = 11.5 μM | 22014559 | ||
| SGC7901 | Cytotoxicity assay | IC50 = 11.5 μM | 24300920 | ||
| HeLa | Antiproliferative activity assay | IC50 = 12.01 μM | 29103873 | ||
| PANC1 | Cytotoxicity assay | IC50 = 12.3 μM | 24300920 | ||
| HepG2 | Antiproliferative activity assay | IC50 = 12.54 μM | 29032031 | ||
| A549 | Antiproliferative activity assay | IC50 = 12.54 μM | 23644219 | ||
| Caki1 | Growth inhibition assay | TGI = 12.59 μM | 28088086 | ||
| OVCAR8 | Growth inhibition assay | TGI = 12.59 μM | 28088086 | ||
| SKOV3 | Antiproliferative activity assay | TGI = 12.6 μM | 27017549 | ||
| ACHN | Antiproliferative activity assay | TGI = 12.6 μM | 27017549 | ||
| OVCAR3 | Antiproliferative activity assay | TGI = 12.6 μM | 27017549 | ||
| OVCAR4 | Antiproliferative activity assay | TGI = 12.6 μM | 27017549 | ||
| HCT15 | Antiproliferative activity assay | TGI = 12.6 μM | 27017549 | ||
| SW620 | Antiproliferative activity assay | TGI = 12.6 μM | 27017549 | ||
| A549/ATCC | Antiproliferative activity assay | TGI = 12.6 μM | 27017549 | ||
| MDA-MB-468 | Cytotoxicity assay | TGI = 13.09 μM | 26590508 | ||
| SK-MEL-28 | Cytotoxicity assay | TGI = 13.39 μM | 26590508 | ||
| A549 | Antiproliferative activity assay | IC50 = 13.64 μM | 26560049 | ||
| A375 | Antiproliferative activity human | IC50 = 13.64 μM | 28242553 | ||
| A375 | Antiproliferative activity assay | IC50 = 13.64 μM | 25462267 | ||
| K562 | Antiproliferative activity assay | IC50 = 13.85 μM | 23644219 | ||
| Hep3B | Cytotoxicity assay | IC50 = 14.08 μM | 21504204 | ||
| T47D | Cytotoxicity assay | TGI = 14.38 μM | 26590508 | ||
| NCI-H226 | Cytotoxicity assay | TGI = 14.42 μM | 26590508 | ||
| MDA-MB-231 | Antiproliferative activity assay | IC50 = 14.62 μM | 24440479 | ||
| MDA-MB-231 | Antiproliferative activity assay | IC50 = 14.62 μM | 23644219 | ||
| NCI-H522 | Cytotoxicity assay | TGI = 14.82 μM | 26590508 | ||
| SK-MEL-2 | Cytotoxicity assay | TGI = 14.85 μM | 26590508 | ||
| OS-RC2 | Cytotoxicity assay | IC50 = 15 μM | 29786436 | ||
| HepG2 | Antiproliferative activity assay | IC50 = 15 μM | 29103873 | ||
| fibroblast | Antiproliferative activity assay | IC50 = 15.1 μM | 18477505 | ||
| HT-29 | Cytotoxicity assay | IC50 = 15.2 μM | 24675135 | ||
| U87MG | Cytotoxicity assay | IC50 = 15.57 μM | 24826815 | ||
| BT549 | Antiproliferative activity assay | LC50 = 15.8 μM | 27017549 | ||
| GES-1 | Cytotoxicity assay | IC50 = 15.8 μM | 23871909 | ||
| SNB19 | Antiproliferative activity assay | TGI = 15.85 μM | 27017549 | ||
| IGROV1 | Antiproliferative activity assay | TGI = 15.9 μM | 27017549 | ||
| HCT116 | Antiproliferative activity assay | LC50 = 15.9 μM | 27017549 | ||
| OS-RC2 | Cytotoxicity assay | IC50 = 16 μM | 23454017 | ||
| A375 | Cytotoxicity assay | IC50 = 16.24 μM | 25461318 | ||
| SKOV3 | Cytotoxicity assay | TGI = 16.36 μM | 26590508 | ||
| WM1361 | Antiproliferative activity assay | IC50 = 16.44 μM | 29602674 | ||
| MKN28 | Antiproliferative activity assay | IC50 = 17 μM | 29032031 | ||
| SMMC7721 | Cytotoxicity assay | IC50 = 17.3 μM | 21504204 | ||
| UACC257 | Cytotoxicity assay | TGI = 17.33 μM | 26590508 | ||
| EAhy926 | Antiangiogenic activity assay | IC50 = 18.52 μM | 29032031 | ||
| SMMC7721 | Cytotoxicity assay | IC50 = 18.7 μM | 24300920 | ||
| Ketr3 | Cytotoxicity assay | IC50 = 18.8 μM | 24675135 | ||
| NCI-H522 | Cytotoxicity assay | IC50 = 19.26 μM | 25461318 | ||
| A549 | Cytotoxicity assay | IC50 = 19.54 μM | 27162123 | ||
| SH-SY5Y | Cytotoxicity assay | IC50 = 19.54 μM | 23871909 | ||
| MGC803 | Antiproliferative activity assay | IC50 = 19.92 μM | 29103873 | ||
| EKVX | Growth inhibition assay | TGI = 19.95 μM | 28088086 | ||
| OVCAR5 | Antiproliferative activity assay | TGI = 20 μM | 27017549 | ||
| UACC62 | Antiproliferative activity assay | LC50 = 20 μM | 27017549 | ||
| UACC257 | Antiproliferative activity assay | LC50 = 20 μM | 27017549 | ||
| MDA-MB-231 | Antiproliferative activity assay | LC50 = 20 μM | 27017549 | ||
| U87 | Cytotoxicity assay | IC50 = 21.07 μM | 20435479 | ||
| EJ | Cytotoxicity assay | IC50 = 22.9 μM | 24300920 | ||
| HCT116 | Antiproliferative activity assay | IC50 = 23.31 μM | 29602674 | ||
| HOP62 | Cytotoxicity assay | TGI = 23.55 μM | 26590508 | ||
| MDA-MB-231 | Cytotoxicity assay | TGI = 23.93 μM | 26590508 | ||
| PC3 | Antiproliferative activity assay | IC50 = 24.2 μM | 24440479 | ||
| PC3 | Antiproliferative activity assay | IC50 = 24.2 μM | 23644219 | ||
| EAhy926 | Antiproliferative activity assay | IC50 = 24.36 μM | 28068599 | ||
| EAhy926 | Anti-angiogenic activity in human | IC50 = 24.36 μM | 29102175 | ||
| U251 | Cytotoxicity assay | IC50 = 24.71 μM | 20435479 | ||
| NIH/3T3 | Antiproliferative activity assay | IC50 = 24.75 μM | 21592628 | ||
| NIH/3T3 | Antiproliferative activity assay | IC50 = 24.75 μM | 22647720 | ||
| DU145 | Cytotoxicity assay | IC50 = 24.91 μM | 27162123 | ||
| DU145 | Cytotoxicity assay | IC50 = 24.91 μM | 23871909 | ||
| U251 | Antiproliferative activity assay | LC50 = 25.1 μM | 27017549 | ||
| M14 | Antiproliferative activity assay | LC50 = 25.1 μM | 27017549 | ||
| NCI-H522 | Antiproliferative activity assay | LC50 = 25.1 μM | 27017549 | ||
| OVCAR4 | Growth inhibition assay | TGI = 25.12 μM | 28088086 | ||
| SH-SY5Y | Cytotoxicity assay | IC50 = 27.71 μM | 27162123 | ||
| A549 | Cytotoxicity assay | IC50 = 27.71 μM | 23871909 | ||
| PLC/PRF/5 | Cytotoxicity assay | IC50 = 29.9 μM | 28109948 | ||
| SK-MEL-5 | Cytotoxicity assay | TGI = 31.26 μM | 26590508 | ||
| A498 | Antiproliferative activity assay | LC50 = 31.6 μM | 27017549 | ||
| SK-MEL-28 | Antiproliferative activity assay | LC50 = 31.6 μM | 27017549 | ||
| KM12 | Antiproliferative activity assay | LC50 = 31.6 μM | 27017549 | ||
| COLO205 | Antiproliferative activity assay | LC50 = 31.6 μM | 27017549 | ||
| NCI-H226 | Antiproliferative activity assay | LC50 = 31.6 μM | 27017549 | ||
| SF295 | Antiproliferative activity assay | LC50 = 31.6 μM | 27017549 | ||
| HOP62 | Antiproliferative activity assay | LC50 = 31.6 μM | 27017549 | ||
| T47D | Antiproliferative activity assay | LC50 = 31.6 μM | 27017549 | ||
| MHCC97L | Cytotoxicity assay | IC50 = 34.41 μM | 28109948 | ||
| MDA-MB-231 | Antiproliferative activity assay | IC50 = 35 μM | 24315192 | ||
| MDA-MB-231 | Cytotoxicity assay | IC50 = 36 μM | 22483592 | ||
| B16-BL6 | Cytotoxicity assay | IC50 = 36.48 μM | 29631788 | ||
| UO31 | Antiproliferative activity assay | LC50 = 39.8 μM | 27017549 | ||
| SN12C | Antiproliferative activity assay | LC50 = 39.8 μM | 27017549 | ||
| RXF393 | Antiproliferative activity assay | LC50 = 39.8 μM | 27017549 | ||
| SNB75 | Antiproliferative activity assay | LC50 = 39.8 μM | 27017549 | ||
| SF268 | Antiproliferative activity assay | LC50 = 39.8 μM | 27017549 | ||
| NCI-H23 | Antiproliferative activity assay | LC50 = 39.8 μM | 27017549 | ||
| HT-29 | Antiproliferative activity assay | LC50 = 39.8 μM | 27017549 | ||
| HOP92 | Antiproliferative activity assay | LC50 = 39.8 μM | 27017549 | ||
| NCI-H460 | Antiproliferative activity assay | LC50 = 39.8 μM | 27017549 | ||
| SW620 | Antiproliferative activity assay | LC50 = 39.8 μM | 27017549 | ||
| MDA-MB-468 | Antiproliferative activity assay | LC50 = 39.8 μM | 27017549 | ||
| MALME-3M | Growth inhibition assay | TGI = 39.81 μM | 28088086 | ||
| 他の多くの細胞株試験データをご覧になる場合はこちらをクリックして下さい | |||||
生物活性
| 製品説明 | Sorafenib is a multikinase inhibitor of Raf-1 and B-Raf with IC50 of 6 nM and 22 nM in cell-free assays, respectively. Sorafenib inhibits VEGFR-2, VEGFR-3, PDGFR-β, Flt-3 and c-KIT with IC50 of 90 nM, 20 nM, 57 nM, 59 nM and 68 nM, respectively. Sorafenib induces autophagy and apoptosis and activates ferroptosis with anti-tumor activity. | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Targets |
|
| In Vitro | ||||
| In vitro | Sorafenib inhibits both wild-type and V599E mutant B-Raf activity with IC50 of 22 nM and 38 nM, respectively. This compound also potently inhibits mVEGFR2 (Flk-1), mVEGFR3, mPDGFRβ, Flt3, and c-Kit with IC50 of 15 nM, 20 nM, 57 nM, 58 nM, and 68 nM, respectively. It weakly inhibits FGFR-1 with IC50 of 580 nM. This chemical is not active against ERK-1, MEK-1, EGFR, HER-2, IGFR-1, c-Met, PKB, PKA, cdk1/cyclinB, PKCα, PKCγ, and pim-1. It markedly inhibits VEGFR2 phosphorylation in NIH 3T3 cells with IC50 of 30 nM, and Flt-3 phosphorylation in HEK-293 cells with IC50 of 20 nM. This agent potently blocks MEK 1/2 and ERK 1/2 phosphorylation in most cell lines but not in A549 or H460 cells, while having no effect on inhibition of the PKB pathway. It inhibits the proliferation of HAoSMC and MDA-MB-231 cells with IC50 of 0.28 μM and 2.6 μM, respectively. In addition to inhibition of the RAF/MEK/ERK signaling pathway, this compound significantly inhibits the phosphorylation of eIF4E and down-regulates Mcl-1 levels in hepatocellular carcinoma (HCC) cells in a MEK/ERK-independent manner. It inhibits the proliferation of PLC/PRF/5 and HepG2 cells with IC50 of 6.3 μM and 4.5 μM, respectively, and leads to the significant induction of apoptosis. | |||
|---|---|---|---|---|
| Kinase Assay | Biochemical assays | |||
| Recombinant baculoviruses expressing Raf-1 (residues 305–648) and B-Raf (residues 409–765) are purified as fusion proteins. Full-length human MEK-1 is generated by PCR and purified as a fusion protein from Escherichia coli lysates. Sorafenib tosylate is added to a mixture of Raf-1 (80 ng), or B-Raf (80 ng) with MEK-1 (1 μg) in assay buffer [20 mM Tris (pH 8.2), 100 mM NaCl, 5 mM MgCl2, and 0.15% β-mercaptoethanol] at a final concentration of 1% DMSO. The Raf kinase assay (final volume of 50 μL) is initiated by adding 25 μL of 10 μM γ[33P]ATP (400 Ci/mol) and incubated at 32 °C for 25 minutes. Phosphorylated MEK-1 is harvested by filtration onto a phosphocellulose mat, and 1% phosphoric acid is used to wash away unbound radioactivity. After drying by microwave heating, a β-plate counter is used to quantify filter-bound radioactivity. Human VEGFR2 (KDR) kinase domain is expressed and purified from Sf9 lysates. Time-resolved fluorescence energy transfer assays for VEGFR2 are performed in 96-well opaque plates in the time-resolved fluorescence energy transfer format. Final reaction conditions are as follows: 1 to 10 μM ATP, 25 nM poly GT-biotin, 2 nM Europium-labeled phospho (p)-Tyr antibody (PY20), 10 nM APC, 1 to 7 nM cytoplasmic kinase domain in final concentrations of 1% DMSO, 50 mM HEPES (pH 7.5), 10 mM MgCl2, 0.1 mM EDTA, 0.015% Brij-35, 0.1 mg/mL BSA, and 0.1% β-mercaptoethanol. Reaction volumes are 100 μL and are initiated by addition of enzyme. Plates are read at both 615 and 665 nM on a Perkin-Elmer VictorV Multilabel counter at ~1.5 to 2.0 hours after reaction initiation. Signal is calculated as a ratio: (665 nm/615 nM) × 10,000 for each well. For IC50 generation, this compound is added before the enzyme initiation. A 50-fold stock plate is made with this compound serially diluted 1:3 in a 50% DMSO/50% distilled water solution. Final concentrations of this chemical range from 10 μM to 4.56 nM in 1% DMSO. | ||||
| 細胞実験 | 細胞株 | MDA-MB-231, and HAoSMC | ||
| 濃度 | Dissolved in DMSO, final concentrations ~10 μM | |||
| 反応時間 | 72 hours | |||
| 実験の流れ | Cells are exposed to increasing concentrations of Sorafenib tosylate for 72 hours. Cell number is quantitated using the Cell TiterGlo ATP Luminescent assay kit. This assay measures the number of viable cells per well by measurement of luminescent signal based on amount of cellular ATP. |
|||
| 実験結果図 | Methods | Biomarkers | 結果図 | PMID |
| Western blot | LC3-I / LC-3II / ATG5 p-STAT3 / STAT3 / Mcl-1 β-catenin / Survivin / Mcl-1 / PTMA pERK / ERK p-PKM2(y105) / PMK2 / Caspase-9 RET(pY1016) / VEGFR2(pY1214) / MEK1(pT292) / ERK(pY204) Cyclin D1 |
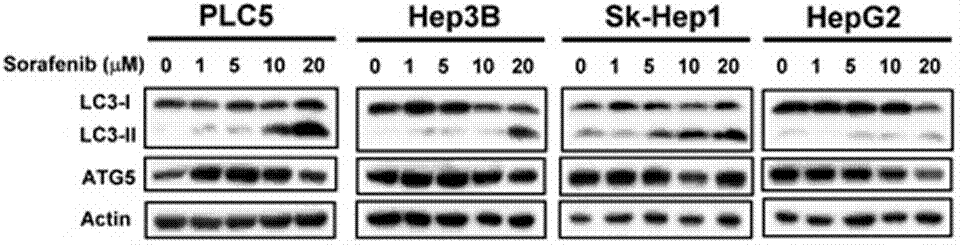
|
23392173 | |
| Growth inhibition assay | Cell viability |
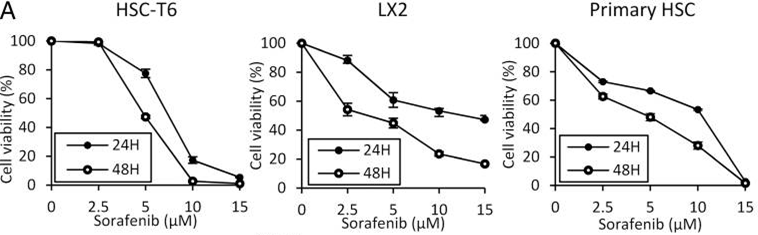
|
26039995 | |
| Immunofluorescence | p65 cytochrome c |
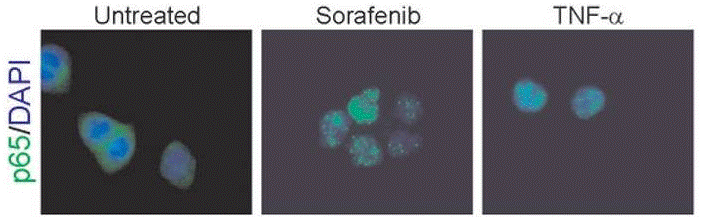
|
22286758 | |
| ELISA | TGF-beta / CD206 Caspase-9 / Caspase-3 |
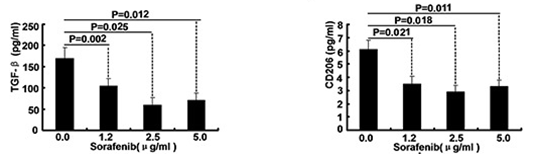
|
26158762 | |
| In Vivo | ||
| In Vivo | Oral administration of Sorafenib (~60 mg/kg) demonstrates broad spectrum, dose-dependent anti-tumor activity against a variety of human tumor xenograft models including MDA-MB-231, Colo-205, HT-29, DLD-1, NCI-H460, and A549, with no evidence of toxicity. In association with the anti-tumor efficacy, this compound treatment potently inhibits MEK 1/2 phosphorylation and pERK 1/2 levels in HT-29 and MDA-MB-231 xenografts but not in Colo-205 xenografts, and significantly suppresses tumor microvessel area (MVA) and microvessel density (MVD) in MDA MB-231, HT-29 and Colo-205 tumor xenografts. This agent produces dose-dependent growth inhibition of PLC/PRF/5 tumor xenografts in SCID mice with TGIs of 49% and 78% at 10 mg/kg and 30 mg/kg, respectively, consistent with the inhibition of ERK and eIF4E phosphorylation, reduction of the microvessel area, and induction of tumor cell apoptosis. It sensitizes bax-/- cells to TRAIL in a dose-dependent manner, through a mechanism involving down-regulating NF-κB mediated Mcl-1 and cIAP2 expression. Combining this compound (30-60 mg/kg) with TRAIL (5 mg/kg) show dramatic efficacy in TRAIL-resistant HCT116 bax-/- and HT29 tumor xenografts. | |
|---|---|---|
| 動物実験 | 動物モデル | Female NCr-nu/nu mice implanted s.c. with MDA-MB-231, Colo-205, HT-29, H460, or A549 cells |
| 投与量 | ~60 mg/kg | |
| 投与経路 | Orally once daily | |
| NCT Number | Recruitment | Conditions | Sponsor/Collaborators | Start Date | Phases |
|---|---|---|---|---|---|
| NCT05068752 | Recruiting | Pancreas Cancer |
HonorHealth Research Institute|Bayer|Genentech Inc. |
October 28 2021 | Phase 2 |
| NCT04763408 | Active not recruiting | Carcinoma Hepatocellular |
Eisai Inc. |
April 9 2021 | -- |
|
化学情報
| 分子量 | 464.82 | 化学式 | C21H16ClF3N4O3 |
| CAS No. | 284461-73-0 | SDF | Download Sorafenib (BAY 43-9006) SDFをダウンロードする |
| Smiles | CNC(=O)C1=NC=CC(=C1)OC2=CC=C(C=C2)NC(=O)NC3=CC(=C(C=C3)Cl)C(F)(F)F | ||
| 保管 | |||
|
In vitro |
DMSO : 92 mg/mL ( (197.92 mM); 吸湿したDMSOは溶解度を減少させます。新しいDMSOをご使用ください。) Water : Insoluble Ethanol : Insoluble |
モル濃度計算器 |
|
in vivo Add solvents to the product individually and in order. |
投与溶液組成計算機 | |||||
実験計算
投与溶液組成計算機(クリア溶液)
ステップ1:実験データを入力してください。(実験操作によるロスを考慮し、動物数を1匹分多くして計算・調製することを推奨します)
mg/kg
g
μL
匹
ステップ2:投与溶媒の組成を入力してください。(ロット毎に適した溶解組成が異なる場合があります。詳細については弊社までお問い合わせください)
% DMSO
%
% Tween 80
% ddH2O
%DMSO
%
計算結果:
投与溶媒濃度: mg/ml;
DMSOストック溶液調製方法: mg 試薬を μL DMSOに溶解する(濃度 mg/mL, 注:濃度が当該ロットのDMSO溶解度を超える場合はご連絡ください。 )
投与溶媒調製方法:Take μL DMSOストック溶液に μL PEG300,を加え、完全溶解後μL Tween 80,を加えて完全溶解させた後 μL ddH2O,を加え完全に溶解させます。
投与溶媒調製方法:μL DMSOストック溶液に μL Corn oil,を加え、完全溶解。
注意:1.ストック溶液に沈殿、混濁などがないことをご確認ください;
2.順番通りに溶剤を加えてください。次のステップに進む前に溶液に沈殿、混濁などがないことを確認してから加えてください。ボルテックス、ソニケーション、水浴加熱など物理的な方法で溶解を早めることは可能です。
技術サポート
ストックの作り方、阻害剤の保管方法、細胞実験や動物実験の際に注意すべき点など、製品を取扱う時に問い合わせが多かった質問に対しては取扱説明書でお答えしています。
他に質問がある場合は、お気軽にお問い合わせください。
* 必須
納期 国内在庫品:受注日の翌日(15時までの受注分) *北海道、九州、沖縄への配送は受注日より2日以上 を要する場合あり 海外在庫品:受注後1〜2週間