
- 阻害剤
- 研究分野別
- PI3K/Akt/mTOR
- Epigenetics
- Methylation
- Immunology & Inflammation
- Protein Tyrosine Kinase
- Angiogenesis
- Apoptosis
- Autophagy
- ER stress & UPR
- JAK/STAT
- MAPK
- Cytoskeletal Signaling
- Cell Cycle
- TGF-beta/Smad
- 化合物ライブラリー
- 抗体
- 新製品
- お問い合わせ
DLAT Antibody (Rabbit mAb) [L23H19]
Catalog No.: F1107
Application:
Reactivity:
-
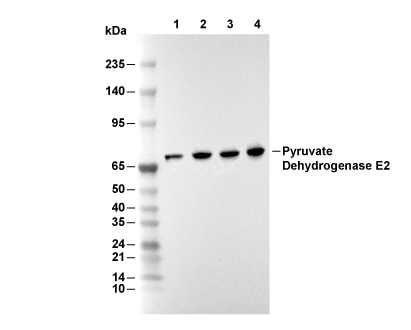 Lane 1: Hela, Lane 2: HepG2, Lane 3: RAW 264.7, Lane 4: C6
Lane 1: Hela, Lane 2: HepG2, Lane 3: RAW 264.7, Lane 4: C6
当該製品は品切れ状态で、メールアドレスをご教示いただければ、お客様に返信いたします。
代表番号: 045-509-1970|電子メール:sales@selleck.co.jp
使用情報
| Dilution |
|---|
|
| Application |
|---|
| WB, IP, IHC, IF, FCM |
| Source |
|---|
| Rabbit Monoclonal Antibody |
| Reactivity |
|---|
| Human |
| Storage Buffer |
|---|
| PBS, pH 7.2+50% Glycerol+0.05% BSA+0.01% NaN3 |
| Storage (from the date of receipt) |
|---|
| -20°C (avoid freeze-thaw cycles), 2 years |
| Predicted MW Observed MW |
|---|
| 69 kDa 68 kDa |
| *なぜ予測分子量と実際の分子量が異なるのか? 下記の原因により、実際の分子量が予測と異なる:タンパク質の翻訳後修飾(リン酸化/糖鎖付加),スプライシングバリアント,イソフォーム,相対的な電荷,ポリマー。 |
| ポジティブコントロール | Human cervical cancer tissue; Human cerebrum tissue; Mouse brain tissue; Mouse kidney tissue; Rat brain tissue; Rat kidney tissue; HeLa cells; A549 cells; HepG2 cells; Jurkat cells; 293T cells; C6 cells; RAW 264.7 cells; PC-12 cells; NIH/3T3 cells |
|---|---|
| ネガティブコントロール |
プロトコール
| WB |
|---|
Experimental Protocol:
Sample preparation
1. Tissue: Lyse the tissue sample by adding an appropriate volume of ice-cold RIPA Lysis Buffer (containing Protease Inhibitor Cocktail),and homogenize the tissue at a low temperature or lyse it by sonication on ice, then incubate on ice for 30 minutes. 2. Adherent cell: Aspirate the culture medium and wash the cells with ice-cold PBS twice. Lyse the cells by adding an appropriate volume of RIPA Lysis Buffer (containing Protease Inhibitor Cocktail) , sonicate to lyse the cells, and incubate on ice for 30 minutes. 3. Suspension cell: Transfer the culture medium to a pre-cooled centrifuge tube. Centrifuge and aspirate the supernatant. Wash the cells with ice-cold PBS twice. Lyse the cells by adding an appropriate volume of RIPA Lysis Buffer (containing Protease Inhibitor Cocktail) , sonicate to lyse the cells, and incubate on ice for 30 minutes. 4. Place the lysate into a pre-cooled microcentrifuge tube. Centrifuge at 4°C for 15 min. Collect the supernatant;
5. Remove a small volume of lysate to determine the protein concentration;
6. Combine the lysate with protein loading buffer. Boil 20 µL sample under 95-100°C for 5 min. Centrifuge for 5 min after cool down on ice.
Electrophoretic separation
1. According to the concentration of extracted protein, load appropriate amount of protein sample and marker onto SDS-PAGE gels for electrophoresis. Recommended separating gel (lower gel) concentration: 10%. Reference Table for Selecting SDS-PAGE Separation Gel Concentrations 2. Power up 80V for 30 minutes. Then the power supply is adjusted (110 V~150 V), the Marker is observed, and the electrophoresis can be stopped when the indicator band of the predyed protein Marker where the protein is located is properly separated. (Note that the current should not be too large when electrophoresis, too large current (more than 150 mA) will cause the temperature to rise, affecting the result of running glue. If high currents cannot be avoided, an ice bath can be used to cool the bath.)
Transfer membrane
1. Take out the converter, soak the clip and consumables in the pre-cooled converter;
2. Activate PVDF membrane with methanol for 1 min and rinse with transfer buffer;
3. Install it in the order of "black edge of clip - sponge - filter paper - filter paper - glue -PVDF membrane - filter paper - filter paper - sponge - white edge of clip"; 4. The protein was electrotransferred to PVDF membrane. ( 0.45 µm PVDF membrane is recommended ) Reference Table for Selecting PVDF Membrane Pore Size Specifications Recommended conditions for wet transfer: 200 mA, 120 min. ( Note that the transfer conditions can be adjusted according to the protein size. For high-molecular-weight proteins, a higher current and longer transfer time are recommended. However, ensure that the transfer tank remains at a low temperature to prevent gel melting.)
Block
1. After electrotransfer, wash the film with TBST at room temperature for 5 minutes;
2. Incubate the film in the blocking solution for 1 hour at room temperature;
3. Wash the film with TBST for 3 times, 5 minutes each time.
Antibody incubation
1. Use 5% skim milk powder to prepare the primary antibody working liquid (recommended dilution ratio for primary antibody 1:1000), gently shake and incubate with the film at 4°C overnight; 2. Wash the film with TBST 3 times, 5 minutes each time;
3. Add the secondary antibody to the blocking solution and incubate with the film gently at room temperature for 1 hour;
4. After incubation, wash the film with TBST 3 times for 5 minutes each time.
Antibody staining
1. Add the prepared ECL luminescent substrate (or select other color developing substrate according to the second antibody) and mix evenly;
2. Incubate with the film for 1 minute, remove excess substrate (keep the film moist), wrap with plastic film, and expose in the imaging system. |
| IF |
|---|
Experimental Protocol:
Sample Preparation
1. Adherent Cells: Place a clean, sterile coverslip in a culture dish. Once the cells grow to near confluence as a monolayer, remove the coverslip for further use.
2. Suspension Cells: Seed the cells onto a clean, sterile slide coated with poly-L-lysine.
3. Frozen Sections: Allow the slide to thaw at room temperature. Wash it with pure water or PBS for 2 times, 3 minutes each time.
4. Paraffin Sections: Deparaffinization and rehydration. Wash the slide with pure water or PBS for 3 times, 3 minutes each time. Then perform antigen retrieval.
Fixation
1. Fix the cell coverslips/spots or tissue sections at room temperature using a fixative such as 4% paraformaldehyde (4% PFA) for 10-15 minutes.
2. Wash the sample with PBS for 3 times, 3 minutes each time.
Permeabilization
1.Add a detergent such as 0.1–0.3% Triton X-100 to the sample and incubate at room temperature for 10–20 minutes.
(Note: This step is only required for intracellular antigens. For antigens expressed on the cell membrane, this step is unnecessary.)
Wash the sample with PBS for 3 times, 3 minutes each time.
Blocking
Add blocking solution and incubate at room temperature for at least 1 hour. (Common blocking solutions include: serum from the same source as the secondary antibody, BSA, or goat serum.)
Note: Ensure the sample remains moist during and after the blocking step to prevent drying, which can lead to high background.
Immunofluorescence Staining (Day 1)
1. Remove the blocking solution and add the diluted primary antibody.
2. Incubate the sample in a humidified chamber at 4°C overnight.
Immunofluorescence Staining (Day 2)
1. Remove the primary antibody and wash with PBST for 3 times, 5 minutes each time.
2. Add the diluted fluorescent secondary antibody and incubate in the dark at 4°C for 1–2 hours.
3. Remove the secondary antibody and wash with PBST for 3 times, 5 minutes each time.
4. Add diluted DAPI and incubate at room temperature in the dark for 5–10 minutes.
5. Wash with PBST for 3 times, 5 minutes each time.
Mounting
1. Mount the sample with an anti-fade mounting medium.
2. Allow the slide to dry at room temperature overnight in the dark.
3. Store the slide in a slide storage box at 4°C, protected from light.
|
| IHC |
|---|
Experimental Protocol:
Deparaffinization/Rehydration
1. Deparaffinize/hydrate sections:
2. Incubate sections in three washes of xylene for 5 min each.
3. Incubate sections in two washes of 100% ethanol for 10 min each.
4. Incubate sections in two washes of 95% ethanol for 10 min each.
5. Wash sections two times in dH2O for 5 min each.
6.Antigen retrieval: For Citrate: Heat slides in a microwave submersed in 1X citrate unmasking solution until boiling is initiated; continue with 10 min at a sub-boiling temperature (95°-98°C). Cool slides on bench top for 30 min.
Staining
1. Wash sections in dH2O three times for 5 min each.
2. Incubate sections in 3% hydrogen peroxide for 10 min.
3. Wash sections in dH2O two times for 5 min each.
4. Wash sections in wash buffer for 5 min.
5. Block each section with 100–400 µl of blocking solution for 1 hr at room temperature.
6. Remove blocking solution and add 100–400 µl primary antibody diluent in to each section. Incubate overnight at 4°C.
7. Remove antibody solution and wash sections with wash buffer three times for 5 min each.
8. Cover section with 1–3 drops HRPas needed. Incubate in a humidified chamber for 30 min at room temperature.
9. Wash sections three times with wash buffer for 5 min each.
10. Add DAB Chromogen Concentrate to DAB Diluent and mix well before use.
11. Apply 100–400 µl DAB to each section and monitor closely. 1–10 min generally provides an acceptable staining intensity.
12. Immerse slides in dH2O.
13. If desired, counterstain sections with hematoxylin.
14. Wash sections in dH2O two times for 5 min each.
15. Dehydrate sections: Incubate sections in 95% ethanol two times for 10 sec each; Repeat in 100% ethanol, incubating sections two times for 10 sec each; Repeat in xylene, incubating sections two times for 10 sec each.
16. Mount sections with coverslips and mounting medium.
|
生物学的記述
| Specificity |
|---|
| DLAT Antibody (Rabbit mAb) [L23H19] detects endogenous levels of total DLAT protein. |
| タンパク質の局在 |
|---|
| ミトコンドリア |
| Uniprot ID |
|---|
| P10515 |
| Clone |
|---|
| L23H19 |
| Synonym(s) |
|---|
| DLTA, DLAT, 70 kDa mitochondrial autoantigen of primary biliary cirrhosis, M2 antigen complex 70 kDa subunit, Pyruvate dehydrogenase complex component E2, PBC, PDC-E2, PDCE2 |
| Background |
|---|
| DLAT (Pyruvate dehydrogenase E2, PDH‑E2, dihydrolipoyl acetyltransferase) forms the inner catalytic core of the mammalian pyruvate dehydrogenase complex (PDC), where it integrates glycolytic carbon flux into mitochondrial acetyl‑CoA production and thereby couples carbohydrate oxidation to the citric acid cycle and downstream lipid biosynthesis. PDH‑E2 belongs to the 2‑oxoacid dehydrogenase family and organizes as a dodecahedral assembly of multiple identical inner‑core domains, which provide the scaffold for peripheral E1 (pyruvate dehydrogenase) and E3 (dihydrolipoamide dehydrogenase) as well as the E3‑binding protein (E3BP) that is specific to mammalian PDC and is embedded into the same core architecture. Each E2 polypeptide contains N‑terminal lipoyl domains and an E1‑binding segment connected by flexible linkers to a C‑terminal inner‑core domain; the inner‑core portion adopts a conserved α/β fold organized into trimers at each three‑fold axis, with these trimers further linked along two‑fold axes into a hollow dodecahedral cage that positions active sites toward internal and external solvent channels. Within each trimer, two neighboring E2 subunits cooperate to form a composite acetyltransferase active site, where a serine residue from one subunit and a histidine from the clockwise partner coordinate binding and positioning of the dihydrolipoyl group and coenzyme A, guiding acetyl transfer from the reduced lipoyllysine arm to CoA during the oxidative decarboxylation sequence initiated by E1. The lipoyllysine “swinging arm” approaches the E2 channel from the outer surface through a narrow passage near helix H1, while CoA accesses the same active site from the inner cavity through a separate portal; shaped electrostatic landscapes at the exterior, channel, and interior surfaces favor productive guidance of lipoylated domains and CoA without nonspecific trapping of substrate or product. A mobile β‑turn between βE and βF at the heart of the active‑site channel lacks ordered density and is positioned to act as a dynamic gate that modulates access and release of lipoyl and CoA ligands, supporting tight coupling of acetyl transfer to upstream decarboxylation and downstream NADH production. Along the two‑fold interface, C‑terminal helices form hydrophobic “knobs” that dock into complementary “holes” on the partner trimer; in human E2, a straight H2 helix and terminal H7 helix remodel this interaction to favor a larger intertrimer angle and thus the dodecahedral rather than cubic geometry seen in other family members, a feature that shapes how E1, E3, and E3BP are arranged and regulated around the core. The same structural surfaces that determine the dodecahedral organization also define how lipoyl domains and regulatory proteins approach the core, so the E2 inner architecture directly influences substrate channeling efficiency, responsiveness to pyruvate dehydrogenase kinases and phosphatases, and the integration of E1 and E3 activities into a single catalytic assembly. A closely related inner‑core domain in E3BP shares high sequence and structural similarity with E2 and can occupy equivalent positions in heterotrimers, while lacking the catalytic histidine; E3BP thereby retains substrate‑binding determinants for the lipoyl arm and CoA but alters the catalytic configuration of one active site within the trimer, creating mixed E2/E3BP trimers that modify local acetyltransferase capacity and the distribution of E3 around the core. Modeling and interface analysis indicate that heterotrimers containing two E2 and one E3BP inner‑core domains are energetically favored, with E3BP‑rich interfaces weaker than E2–E2 contacts; this arrangement supports a core in which E3BP subunits are inserted in a limited, patterned manner that preserves overall dodecahedral geometry while tuning E3 recruitment and local catalytic environment. Through this modular organization, PDH‑E2 not only catalyzes acetyl transfer but also determines the spatial pattern of catalytic centers and E3 docking, thereby shaping flux through pyruvate oxidation and influencing how PDC responds to changes in nutrient availability, redox balance, and hormonal input that signal through pyruvate dehydrogenase kinases and phosphatases. The irreversible conversion of pyruvate to acetyl‑CoA, supported by the E2 core, constitutes a major control point in mammalian energy metabolism, and reduction in overall PDC function due to defects in E2, E3BP, or their assembly leads to impaired oxidative metabolism, accumulation of pyruvate and lactate, and neurological dysfunction linked to lactic acidosis and other metabolic disorders. |
| References |
|---|
技術サポート
ストックの作り方、阻害剤の保管方法、細胞実験や動物実験の際に注意すべき点など、製品を取扱う時に問い合わせが多かった質問に対しては取扱説明書でお答えしています。
他に質問がある場合は、お気軽にお問い合わせください。
* 必須
納期 国内在庫品:受注日の翌日(15時までの受注分) *北海道、九州、沖縄への配送は受注日より2日以上 を要する場合あり 海外在庫品:受注後1〜2週間