
- 阻害剤
- 研究分野別
- PI3K/Akt/mTOR
- Epigenetics
- Methylation
- Immunology & Inflammation
- Protein Tyrosine Kinase
- Angiogenesis
- Apoptosis
- Autophagy
- ER stress & UPR
- JAK/STAT
- MAPK
- Cytoskeletal Signaling
- Cell Cycle
- TGF-beta/Smad
- 化合物ライブラリー
- 抗体
- 新製品
- お問い合わせ
Aurora Kinase
Aurora Kinase製品
- All (40)
- Aurora Kinase阻害剤 (40)
- 新製品
| 製品コード | 製品名称 | 製品説明 | 文献中Selleckの製品使用例 | お客様のフィードバック |
|---|---|---|---|---|
| S1460 | SP600125 | SP600125(Nsc75890)は、JNK1、JNK2、JNK3に対する広範なJNK阻害剤であり、細胞フリーアッセイでのIC50はそれぞれ40nM、40nM、90nMです。MKK4に対して10倍、MKK3、MKK6、PKB、PKCαに対して25倍、ERK2、p38、Chk1、EGFRなどに対して100倍高い選択性を示します。SP600125は、Aurora kinase A、FLT3、TRKAを含むserine/threonine kinasesの広範な阻害剤でもあり、IC50はそれぞれ60nM、90nM、70nMです。SP600125はautophagyを阻害し、apoptosisを活性化します。 |

|
|
| S1133 | Alisertib (MLN8237) | Alisertib (MLN8237)は、無細胞アッセイにおいてIC50が1.2 nMの選択的Aurora A阻害剤です。Aurora BよりもAurora Aに対して200倍以上の選択性を示します。Alisertibは細胞周期停止、apoptosis、およびautophagyを誘導します。現在治験第3相です。 |
-S113303W0120130926.gif)
|
|
| S1147 | Barasertib-HQPA (AZD2811) | Defosbarasertib (AZD1152-HQPA, AZD2811, INH-34, Barasertib-HQPA) は、高選択的なAurora B阻害剤で、in vitroアッセイにおけるIC50値は0.37 nMです。Aurora AよりもAurora Bに対して約3700倍高い選択性を示します。第1相。 |

|
|
| S1048 | Tozasertib (VX-680, MK-0457) | Tozasertib (VX-680)は、Aurora Aに対するKiappがセルフリーアッセイで0.6 nMのパンAurora阻害剤であり、Aurora B/Aurora Cに対する効力は低く、他の55のキナーゼよりもAurora Aに対して100倍選択的です。唯一の例外は、Kiが30 nMでTozasertibによって阻害されるFms-related tyrosine kinase-3 (FLT-3)とBCR-ABL tyrosine kinaseです。Tozasertibはapoptosisとautophagyを誘導します。フェーズ2。 |
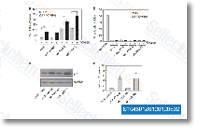
|
|
| S1103 | ZM 447439 | ZM 447439は、Aurora AとAurora Bに対する選択的かつATP競合的阻害剤であり、それぞれ110 nMと130 nMのIC50を示します。MEK1、Src、LckよりもAurora A/Bに対して8倍以上の選択性があり、CDK1/2/4、Plk1、Chk1などに対してはほとんど影響を与えません。 |

|
|
| S1100 | MLN8054 | MLN8054は、Sf9昆虫細胞においてIC50が4nMの、強力かつ選択的なAurora A阻害剤です。Aurora BよりもAurora Aに対して40倍以上の選択性を示します。フェーズ1。 |
|
|
| S1529 | Hesperadin | Hesperadinは、セルフリーアッセイにおいて250 nMのIC50でAurora Bを強力に阻害します。AMPK、Lck、MKK1、MAPKAP-K1、CHK1、およびPHKの活性を著しく低下させますが、in vivoではMKK1活性を阻害しません。 |

|
|
| S1107 | Danusertib (PHA-739358) | Danusertib (PHA-739358)は、細胞を含まないアッセイでAurora A/B/Cに対するIC50が13 nM/79 nM/61 nMのAurora Kinase阻害剤であり、Abl、TrkA、c-RET、FGFR1に対して中程度の効力を持ち、Lck、VEGFR2/3、c-Kit、CDK2などに対してはより低い効力を持つ。Danusertibはapoptosis、細胞周期停止、およびautophagyを誘導する。フェーズ2。 |

|
|
| S2770 | MK-5108 | MK-5108は、無細胞アッセイにおいて0.064 nMのIC50を持つ高選択的なAurora A阻害剤であり、Aurora B/Cに対しては220倍および190倍の選択性を示しますが、TrkAに対しては100倍未満の選択性で阻害します。MK-5108(VX-689)はautophagyを誘導します。フェーズ1。 |
-S277001W0220130927.gif)
|
|
| S1451 | TCS7010 (Aurora A Inhibitor I) | TCS7010 (Aurora A Inhibitor I) は、3.4 nMのIC50値を示す、新規かつ強力なAurora Aの選択的阻害剤です。Aurora Bと比較して、Aurora Aに対する選択性は1000倍高いです。Aurora A Inhibitor I (TC-S 7010) は、ROSを介したUPRシグナル伝達経路を介してapoptosisを誘発します。 |

|
|
| S1134 | AT9283 | AT9283は、in vitroアッセイにおいてIC50が1.2 nM/1.1 nMの強力なJAK2/3阻害剤であり、Aurora A/BおよびAbl1(T315I)にも強力に作用します。 |

|
|
| S1249 | JNJ-7706621 | JNJ-7706621は、IC50が9 nM/4 nMでCDK1/2に最高の効力を持つパン-CDK阻害剤であり、セルフリーアッセイではCDK3/4/6よりもCDK1/2に対して>6倍の選択性を示します。また、Aurora A/Bを強力に阻害し、Plk1およびWee1には活性がありません。 |
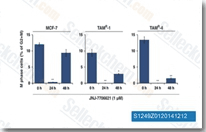
|
|
| S2719 | AMG-900 | AMG 900 は、Aurora A/B/C に対する強力かつ高選択的な pan-Aurora kinases 阻害剤であり、IC50 は 5 nM/4 nM /1 nM です。p38α、Tyk2、JNK2、Met、Tie2 よりも Aurora kinases に対して 10 倍以上の選択性を示します。フェーズ 1。 |
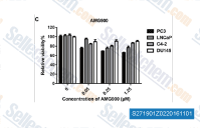
|
|
| S2744 | CCT137690 | CCT137690 は、Aurora A、Aurora B、Aurora C の高選択的阻害剤であり、IC50 はそれぞれ 15 nM、25 nM、19 nM です。hERG イオンチャネルにはほとんど影響を与えません。 |

|
|
| S7588 | Reversine | Reversineは、ヒトA3アデノシン受容体の強力なアンタゴニストであり、Ki値は0.66 μMです。また、パンオーロラA/B/Cキナーゼ阻害剤でもあり、それぞれのIC50値は400 nM/500 nM/400 nMです。幹細胞の脱分化にも使用されます。 |

|
|
| S1454 | PHA-680632 | PHA-680632は、Aurora A、Aurora B、およびAurora Cの強力な阻害剤であり、それぞれのIC50は27 nM、135 nM、120 nMです。FGFR1、FLT3、LCK、PLK1、STLK2、VEGFR2/3に対するIC50は10~200倍高いです。 |
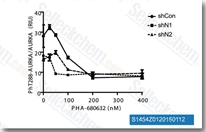
|
|
| S1181 | ENMD-2076 | ENMD-2076は、Aurora AおよびFlt3に対し選択的な活性を持ち、IC50値はそれぞれ14 nMおよび1.86 nMです。Aurora BよりもAurora Aに対して25倍選択的であり、RET、SRC、NTRK1/TRKA、CSF1R/FMS、VEGFR2/KDR、FGFR、PDGFRαに対してはより低効力です。ENMD-2076は、広範囲のヒト固形腫瘍および造血器癌細胞株の増殖を0.025〜0.7 μMのIC50で阻害し、apoptosisおよびG2/M期停止を誘導します。フェーズ2。 |

|
|
| S2740 | GSK1070916 | GSK1070916は、Aurora B/Cの可逆的ATP競合阻害剤であり、IC50は3.5 nM/6.5 nMです。これは、密接に関連するAurora A-TPX2複合体に対して100倍以上の選択性を示します。フェーズ1。 |

|
|
| S2158 | KW-2449 | KW-2449は、IC50が6.6 nMで主にFLT3を標的とする多標的阻害剤であり、FGFR1、Bcr-Abl、Aurora Aに対して中程度の効力があります。PDGFRβ、IGF-1R、EGFRにはほとんど影響がありません。第1相。 |

|
|
| S1154 | SNS-314 | SNS-314メシル酸塩は、Aurora A、Aurora B、Aurora Cの強力かつ選択的な阻害剤であり、それぞれのIC50値は9 nM、31 nM、3 nMです。Trk A/B、Flt4、Fms、Axl、c-Raf、DDR2に対する効力はそれほど高くありません。第1相。 |

|
|
| S1171 | CYC116 | CYC116はAurora A/Bの強力な阻害剤であり、Kiは8.0 nM/9.2 nMで、VEGFR2(Kiは44 nM)に対する効力は低く、CDKに比べて50倍高い効力を持ち、PKA、Akt/PKB、PKCに対しては活性がなく、GSK-3α/β、CK2、Plk1、SAPK2Aには影響を与えません。フェーズ1。 |

|
|
| S2718 | TAK-901 | TAK-901は、IC50が21nM/15nMであるAurora A/Bの新規阻害剤です。細胞性JAK2、c-Src、またはAblの強力な阻害剤ではありません。フェーズ1。 | ||
| S8782 | LY3295668 | LY3295668 (AK-01) は、Aurora A キナーゼの強力で経口活性のある特異的阻害剤であり、AURKA および AURKB に対する Ki はそれぞれ 0.8 nM および 1038 nM です。 | ||
| S7843 | BI-847325 | BI-847325は、経口投与可能で選択的なデュアルMEK/Auroraキナーゼ阻害剤であり、Xenopus laevis Aurora B、ヒトAurora A、Aurora C、およびヒトMEK1とMEK2に対するIC50値はそれぞれ3 nM、25 nM、15 nM、25 nM、および4 nMです。第1相。 | ||
| S1051 | AZD1152(Barasertib) | AZD1152は、Aurora Bの選択的阻害剤であり、Kiは0.36 nMです。 | ||
| S1519 | CCT129202 | CCT129202は、Aurora A、Aurora B、Aurora Cに対するATP競合型pan-Aurora阻害剤であり、それぞれのIC50は0.042 µM、0.198 µM、0.227 µMです。FGFR3、GSK3β、PDGFRβなどに対する効力は低いです。 |

|
|
| S8699 | SNS-314 Mesylate | SNS-314 Mesylateは、Aurora A、Aurora B、Aurora Cに対する強力かつ選択的な阻害剤であり、IC50値はそれぞれ9 nM、31 nM、3 nMです。また、Trk A/B、Flt4、Fms、Axl、c-Raf、DDR2に対しては、より低い効力しか持ちません。 | ||
| S7065 | MK-8745 | MK-8745は、Aurora Aの強力かつ選択的阻害剤であり、IC50は0.6 nMで、Aurora BよりもAurora Aに対して450倍以上の選択性を示します。 | ||
| S2018 | ENMD-2076 L-(+)-Tartaric acid | ENMD-2076 L-(+)-酒石酸は、ENMD-2076の酒石酸であり、Aurora AおよびFlt3に対し、IC50が14 nMおよび1.86 nMの選択的活性を示し、Aurora BよりもAurora Aに対して25倍選択的であり、VEGFR2/KDR、VEGFR3、FGFR1、FGFR2、PDGFRαに対する効力は低い。第2相。 |
|
|
| S9658 | SP-96 | SP-96は、IC50が0.316 nMの強力で選択的な非ATP競合性のAurora B阻害剤です。SP-96は、トリプルネガティブ乳がん(TNBC)の研究に使用できます。 | ||
| S6214 | H-1152 dihydrochloride | H-1152 dihydrochloride(2HCl)は、膜透過性のある選択的なRho関連プロテインキナーゼ(ROCK)阻害剤です。H-1152は、ROCK2、PKA、PKC、PKG、AuroraA、CaMK2をそれぞれIC50値0.0120 µM、3.03 µM、5.68 µM、0.360 µM、0.745 µM、0.180 µMで阻害します。 | ||
| E1651 | CD532 | CD532 は、45 nM の IC50 を持つ強力な Aurora A キナーゼ阻害剤です。Aurora-A の本来のコンフォメーションを破壊し、N-Myc 駆動型癌における N-Myc の分解を促進し、抗増殖効果も示します。 | ||
| S9741 | TAS-119 | TAS-119は、Aurora Aを含むAurora Bに対し、それぞれIC50が1.0 nMおよび95 nMである、強力で選択的かつ経口活性型の阻害剤です。抗腫瘍活性を示します。 | ||
| E0338 | AKI603 | AKI-603は、BCR-ABL-T315I変異によって媒介される抵抗性を克服するために開発されたAuroraキナーゼA(AurA)の阻害剤です。 | ||
| E0342 | Aurora kinase Inhibitor II | Aurora kinase Inhibitor IIは、強力かつ選択的なATP競合阻害剤であるアニリノキナゾリンであり、細胞透過性があり、細胞周期、特に細胞分裂の調節に関与しています。 | ||
| E0616 | Chiauranib | Chiauranib(CS2164)は、複数のキナーゼターゲットであるオーロラBキナーゼ(AURKB)、コロニー刺激因子1受容体(CSF1R)、血管内皮増殖因子受容体(VEGFR)/血小板由来増殖因子受容体(PDGFR)/c-Kitを選択的に阻害することで、腫瘍細胞の急速な増殖を抑制し、抗腫瘍免疫を増強し、腫瘍血管新生を阻害し、抗腫瘍効果を発揮します。 | ||
| E5904 | 6K465 | 6K465は、c-MYCおよびN-MYCオンコプロテインのレベルを低下させるAurora Aのピリミジンベース阻害剤です。これは説得力のある抗腫瘍効果を示します。 | ||
| S2931 | Aurora Kinase Inhibitor III | Aurora Kinase Inhibitor IIIは、IC50が42nMのAurora A kinaseの強力な阻害剤であり、BMX、BTK、IGF-1R、c-Src、TRKB、SYK、およびEGFR(IC50sはそれぞれ386、3,550、591、1,980、2,510、887、および>10,000nM)に対してAurora Aの高い選択性を示します。 | ||
| S1272 | XL228 | XL228は、野生型ABLキナーゼ、ABL T315I、Aurora A、IGF-1R、SRC、およびLYNに対して、それぞれ5 nM、1.4 nM、3.1 nM、1.6 nM、6.1 nM、2 nMのIC50値を持つプロテインキナーゼ阻害剤です。 | ||
| E1435 | Tinengotinib | Tinengotinib (TT-00420) は、IC50値がそれぞれ1.2/3.3 nMでAurora A/Bを強力に阻害する新規多重キナーゼ阻害剤です。また、FGFR1/2/3、VEGFR1、JAK1/2、およびCSF1Rに対して強力な阻害活性を示し、トリプルネガティブ乳がん(TNBC)のいくつかの主要なシグナル伝達経路の治療に使用できます。 | ||
| S1460 | SP600125 | SP600125(Nsc75890)は、JNK1、JNK2、JNK3に対する広範なJNK阻害剤であり、細胞フリーアッセイでのIC50はそれぞれ40nM、40nM、90nMです。MKK4に対して10倍、MKK3、MKK6、PKB、PKCαに対して25倍、ERK2、p38、Chk1、EGFRなどに対して100倍高い選択性を示します。SP600125は、Aurora kinase A、FLT3、TRKAを含むserine/threonine kinasesの広範な阻害剤でもあり、IC50はそれぞれ60nM、90nM、70nMです。SP600125はautophagyを阻害し、apoptosisを活性化します。 |

|
|
| S1133 | Alisertib (MLN8237) | Alisertib (MLN8237)は、無細胞アッセイにおいてIC50が1.2 nMの選択的Aurora A阻害剤です。Aurora BよりもAurora Aに対して200倍以上の選択性を示します。Alisertibは細胞周期停止、apoptosis、およびautophagyを誘導します。現在治験第3相です。 |
-S113303W0120130926.gif)
|
|
| S1147 | Barasertib-HQPA (AZD2811) | Defosbarasertib (AZD1152-HQPA, AZD2811, INH-34, Barasertib-HQPA) は、高選択的なAurora B阻害剤で、in vitroアッセイにおけるIC50値は0.37 nMです。Aurora AよりもAurora Bに対して約3700倍高い選択性を示します。第1相。 |

|
|
| S1048 | Tozasertib (VX-680, MK-0457) | Tozasertib (VX-680)は、Aurora Aに対するKiappがセルフリーアッセイで0.6 nMのパンAurora阻害剤であり、Aurora B/Aurora Cに対する効力は低く、他の55のキナーゼよりもAurora Aに対して100倍選択的です。唯一の例外は、Kiが30 nMでTozasertibによって阻害されるFms-related tyrosine kinase-3 (FLT-3)とBCR-ABL tyrosine kinaseです。Tozasertibはapoptosisとautophagyを誘導します。フェーズ2。 |

|
|
| S1103 | ZM 447439 | ZM 447439は、Aurora AとAurora Bに対する選択的かつATP競合的阻害剤であり、それぞれ110 nMと130 nMのIC50を示します。MEK1、Src、LckよりもAurora A/Bに対して8倍以上の選択性があり、CDK1/2/4、Plk1、Chk1などに対してはほとんど影響を与えません。 |

|
|
| S1100 | MLN8054 | MLN8054は、Sf9昆虫細胞においてIC50が4nMの、強力かつ選択的なAurora A阻害剤です。Aurora BよりもAurora Aに対して40倍以上の選択性を示します。フェーズ1。 |

|
|
| S1529 | Hesperadin | Hesperadinは、セルフリーアッセイにおいて250 nMのIC50でAurora Bを強力に阻害します。AMPK、Lck、MKK1、MAPKAP-K1、CHK1、およびPHKの活性を著しく低下させますが、in vivoではMKK1活性を阻害しません。 |

|
|
| S1107 | Danusertib (PHA-739358) | Danusertib (PHA-739358)は、細胞を含まないアッセイでAurora A/B/Cに対するIC50が13 nM/79 nM/61 nMのAurora Kinase阻害剤であり、Abl、TrkA、c-RET、FGFR1に対して中程度の効力を持ち、Lck、VEGFR2/3、c-Kit、CDK2などに対してはより低い効力を持つ。Danusertibはapoptosis、細胞周期停止、およびautophagyを誘導する。フェーズ2。 |

|
|
| S2770 | MK-5108 | MK-5108は、無細胞アッセイにおいて0.064 nMのIC50を持つ高選択的なAurora A阻害剤であり、Aurora B/Cに対しては220倍および190倍の選択性を示しますが、TrkAに対しては100倍未満の選択性で阻害します。MK-5108(VX-689)はautophagyを誘導します。フェーズ1。 |
-S277001W0220130927.gif)
|
|
| S1451 | TCS7010 (Aurora A Inhibitor I) | TCS7010 (Aurora A Inhibitor I) は、3.4 nMのIC50値を示す、新規かつ強力なAurora Aの選択的阻害剤です。Aurora Bと比較して、Aurora Aに対する選択性は1000倍高いです。Aurora A Inhibitor I (TC-S 7010) は、ROSを介したUPRシグナル伝達経路を介してapoptosisを誘発します。 |

|
|
| S1134 | AT9283 | AT9283は、in vitroアッセイにおいてIC50が1.2 nM/1.1 nMの強力なJAK2/3阻害剤であり、Aurora A/BおよびAbl1(T315I)にも強力に作用します。 |

|
|
| S1249 | JNJ-7706621 | JNJ-7706621は、IC50が9 nM/4 nMでCDK1/2に最高の効力を持つパン-CDK阻害剤であり、セルフリーアッセイではCDK3/4/6よりもCDK1/2に対して>6倍の選択性を示します。また、Aurora A/Bを強力に阻害し、Plk1およびWee1には活性がありません。 |

|
|
| S2719 | AMG-900 | AMG 900 は、Aurora A/B/C に対する強力かつ高選択的な pan-Aurora kinases 阻害剤であり、IC50 は 5 nM/4 nM /1 nM です。p38α、Tyk2、JNK2、Met、Tie2 よりも Aurora kinases に対して 10 倍以上の選択性を示します。フェーズ 1。 |

|
|
| S2744 | CCT137690 | CCT137690 は、Aurora A、Aurora B、Aurora C の高選択的阻害剤であり、IC50 はそれぞれ 15 nM、25 nM、19 nM です。hERG イオンチャネルにはほとんど影響を与えません。 |

|
|
| S7588 | Reversine | Reversineは、ヒトA3アデノシン受容体の強力なアンタゴニストであり、Ki値は0.66 μMです。また、パンオーロラA/B/Cキナーゼ阻害剤でもあり、それぞれのIC50値は400 nM/500 nM/400 nMです。幹細胞の脱分化にも使用されます。 |

|
|
| S1454 | PHA-680632 | PHA-680632は、Aurora A、Aurora B、およびAurora Cの強力な阻害剤であり、それぞれのIC50は27 nM、135 nM、120 nMです。FGFR1、FLT3、LCK、PLK1、STLK2、VEGFR2/3に対するIC50は10~200倍高いです。 |

|
|
| S1181 | ENMD-2076 | ENMD-2076は、Aurora AおよびFlt3に対し選択的な活性を持ち、IC50値はそれぞれ14 nMおよび1.86 nMです。Aurora BよりもAurora Aに対して25倍選択的であり、RET、SRC、NTRK1/TRKA、CSF1R/FMS、VEGFR2/KDR、FGFR、PDGFRαに対してはより低効力です。ENMD-2076は、広範囲のヒト固形腫瘍および造血器癌細胞株の増殖を0.025〜0.7 μMのIC50で阻害し、apoptosisおよびG2/M期停止を誘導します。フェーズ2。 |

|
|
| S2740 | GSK1070916 | GSK1070916は、Aurora B/Cの可逆的ATP競合阻害剤であり、IC50は3.5 nM/6.5 nMです。これは、密接に関連するAurora A-TPX2複合体に対して100倍以上の選択性を示します。フェーズ1。 |

|
|
| S2158 | KW-2449 | KW-2449は、IC50が6.6 nMで主にFLT3を標的とする多標的阻害剤であり、FGFR1、Bcr-Abl、Aurora Aに対して中程度の効力があります。PDGFRβ、IGF-1R、EGFRにはほとんど影響がありません。第1相。 |

|
|
| S1154 | SNS-314 | SNS-314メシル酸塩は、Aurora A、Aurora B、Aurora Cの強力かつ選択的な阻害剤であり、それぞれのIC50値は9 nM、31 nM、3 nMです。Trk A/B、Flt4、Fms、Axl、c-Raf、DDR2に対する効力はそれほど高くありません。第1相。 |

|
|
| S1171 | CYC116 | CYC116はAurora A/Bの強力な阻害剤であり、Kiは8.0 nM/9.2 nMで、VEGFR2(Kiは44 nM)に対する効力は低く、CDKに比べて50倍高い効力を持ち、PKA、Akt/PKB、PKCに対しては活性がなく、GSK-3α/β、CK2、Plk1、SAPK2Aには影響を与えません。フェーズ1。 |

|
|
| S2718 | TAK-901 | TAK-901は、IC50が21nM/15nMであるAurora A/Bの新規阻害剤です。細胞性JAK2、c-Src、またはAblの強力な阻害剤ではありません。フェーズ1。 | ||
| S8782 | LY3295668 | LY3295668 (AK-01) は、Aurora A キナーゼの強力で経口活性のある特異的阻害剤であり、AURKA および AURKB に対する Ki はそれぞれ 0.8 nM および 1038 nM です。 | ||
| S7843 | BI-847325 | BI-847325は、経口投与可能で選択的なデュアルMEK/Auroraキナーゼ阻害剤であり、Xenopus laevis Aurora B、ヒトAurora A、Aurora C、およびヒトMEK1とMEK2に対するIC50値はそれぞれ3 nM、25 nM、15 nM、25 nM、および4 nMです。第1相。 | ||
| S1051 | AZD1152(Barasertib) | AZD1152は、Aurora Bの選択的阻害剤であり、Kiは0.36 nMです。 | ||
| S1519 | CCT129202 | CCT129202は、Aurora A、Aurora B、Aurora Cに対するATP競合型pan-Aurora阻害剤であり、それぞれのIC50は0.042 µM、0.198 µM、0.227 µMです。FGFR3、GSK3β、PDGFRβなどに対する効力は低いです。 |

|
|
| S8699 | SNS-314 Mesylate | SNS-314 Mesylateは、Aurora A、Aurora B、Aurora Cに対する強力かつ選択的な阻害剤であり、IC50値はそれぞれ9 nM、31 nM、3 nMです。また、Trk A/B、Flt4、Fms、Axl、c-Raf、DDR2に対しては、より低い効力しか持ちません。 | ||
| S7065 | MK-8745 | MK-8745は、Aurora Aの強力かつ選択的阻害剤であり、IC50は0.6 nMで、Aurora BよりもAurora Aに対して450倍以上の選択性を示します。 | ||
| S2018 | ENMD-2076 L-(+)-Tartaric acid | ENMD-2076 L-(+)-酒石酸は、ENMD-2076の酒石酸であり、Aurora AおよびFlt3に対し、IC50が14 nMおよび1.86 nMの選択的活性を示し、Aurora BよりもAurora Aに対して25倍選択的であり、VEGFR2/KDR、VEGFR3、FGFR1、FGFR2、PDGFRαに対する効力は低い。第2相。 |
|
|
| S9658 | SP-96 | SP-96は、IC50が0.316 nMの強力で選択的な非ATP競合性のAurora B阻害剤です。SP-96は、トリプルネガティブ乳がん(TNBC)の研究に使用できます。 | ||
| S6214 | H-1152 dihydrochloride | H-1152 dihydrochloride(2HCl)は、膜透過性のある選択的なRho関連プロテインキナーゼ(ROCK)阻害剤です。H-1152は、ROCK2、PKA、PKC、PKG、AuroraA、CaMK2をそれぞれIC50値0.0120 µM、3.03 µM、5.68 µM、0.360 µM、0.745 µM、0.180 µMで阻害します。 | ||
| E1651 | CD532 | CD532 は、45 nM の IC50 を持つ強力な Aurora A キナーゼ阻害剤です。Aurora-A の本来のコンフォメーションを破壊し、N-Myc 駆動型癌における N-Myc の分解を促進し、抗増殖効果も示します。 | ||
| S9741 | TAS-119 | TAS-119は、Aurora Aを含むAurora Bに対し、それぞれIC50が1.0 nMおよび95 nMである、強力で選択的かつ経口活性型の阻害剤です。抗腫瘍活性を示します。 | ||
| E0338 | AKI603 | AKI-603は、BCR-ABL-T315I変異によって媒介される抵抗性を克服するために開発されたAuroraキナーゼA(AurA)の阻害剤です。 | ||
| E0342 | Aurora kinase Inhibitor II | Aurora kinase Inhibitor IIは、強力かつ選択的なATP競合阻害剤であるアニリノキナゾリンであり、細胞透過性があり、細胞周期、特に細胞分裂の調節に関与しています。 | ||
| E0616 | Chiauranib | Chiauranib(CS2164)は、複数のキナーゼターゲットであるオーロラBキナーゼ(AURKB)、コロニー刺激因子1受容体(CSF1R)、血管内皮増殖因子受容体(VEGFR)/血小板由来増殖因子受容体(PDGFR)/c-Kitを選択的に阻害することで、腫瘍細胞の急速な増殖を抑制し、抗腫瘍免疫を増強し、腫瘍血管新生を阻害し、抗腫瘍効果を発揮します。 | ||
| E5904 | 6K465 | 6K465は、c-MYCおよびN-MYCオンコプロテインのレベルを低下させるAurora Aのピリミジンベース阻害剤です。これは説得力のある抗腫瘍効果を示します。 | ||
| S2931 | Aurora Kinase Inhibitor III | Aurora Kinase Inhibitor IIIは、IC50が42nMのAurora A kinaseの強力な阻害剤であり、BMX、BTK、IGF-1R、c-Src、TRKB、SYK、およびEGFR(IC50sはそれぞれ386、3,550、591、1,980、2,510、887、および>10,000nM)に対してAurora Aの高い選択性を示します。 | ||
| S1272 | XL228 | XL228は、野生型ABLキナーゼ、ABL T315I、Aurora A、IGF-1R、SRC、およびLYNに対して、それぞれ5 nM、1.4 nM、3.1 nM、1.6 nM、6.1 nM、2 nMのIC50値を持つプロテインキナーゼ阻害剤です。 | ||
| E1435 | Tinengotinib | Tinengotinib (TT-00420) は、IC50値がそれぞれ1.2/3.3 nMでAurora A/Bを強力に阻害する新規多重キナーゼ阻害剤です。また、FGFR1/2/3、VEGFR1、JAK1/2、およびCSF1Rに対して強力な阻害活性を示し、トリプルネガティブ乳がん(TNBC)のいくつかの主要なシグナル伝達経路の治療に使用できます。 |
Aurora Kinaseシグナル伝達経路
納期 国内在庫品:受注日の翌日(15時までの受注分) *北海道、九州、沖縄への配送は受注日より2日以上 を要する場合あり 海外在庫品:受注後1〜2週間