
- 阻害剤
- 研究分野別
- PI3K/Akt/mTOR
- Epigenetics
- Methylation
- Immunology & Inflammation
- Protein Tyrosine Kinase
- Angiogenesis
- Apoptosis
- Autophagy
- ER stress & UPR
- JAK/STAT
- MAPK
- Cytoskeletal Signaling
- Cell Cycle
- TGF-beta/Smad
- 化合物ライブラリー
- 抗体
- 新製品
- お問い合わせ
FLT3
亜型選択性的な製品
FLT3製品
- All (53)
- FLT3阻害剤 (53)
- 新製品
| 製品コード | 製品名称 | 製品説明 | 文献中Selleckの製品使用例 | お客様のフィードバック |
|---|---|---|---|---|
| S1460 | SP600125 | SP600125(Nsc75890)は、JNK1、JNK2、JNK3に対する広範なJNK阻害剤であり、細胞フリーアッセイでのIC50はそれぞれ40nM、40nM、90nMです。MKK4に対して10倍、MKK3、MKK6、PKB、PKCαに対して25倍、ERK2、p38、Chk1、EGFRなどに対して100倍高い選択性を示します。SP600125は、Aurora kinase A、FLT3、TRKAを含むserine/threonine kinasesの広範な阻害剤でもあり、IC50はそれぞれ60nM、90nM、70nMです。SP600125はautophagyを阻害し、apoptosisを活性化します。 |

|
|
| S7397 | Sorafenib (BAY 43-9006) | Sorafenib は、細胞を含まないアッセイで Raf-1 および B-Raf のマルチキナーゼ阻害剤であり、それぞれ 6 nM および 22 nM の IC50 を示します。Sorafenib は、VEGFR-2、VEGFR-3、PDGFR-β、Flt-3 および c-KIT をそれぞれ 90 nM、20 nM、57 nM、59 nM および 68 nM の IC50 で阻害します。Sorafenib は、抗腫瘍活性を持つ autophagy と apoptosis を誘導し、ferroptosis を活性化します。 |
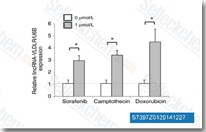
|
|
| S1040 | Sorafenib Tosylate (BAY 43-9006) | Sorafenib tosylateは、無細胞アッセイにおいてIC50がそれぞれ6 nMおよび22 nMのRaf-1およびB-Rafのマルチキナーゼ阻害剤です。Sorafenib Tosylateは、VEGFR-2、VEGFR-3、PDGFR-β、Flt-3、およびc-KITをそれぞれ90 nM、20 nM、57 nM、59 nM、および68 nMのIC50で阻害します。Sorafenib Tosylateは、抗腫瘍活性を伴うautophagyとapoptosisを誘導し、ferroptosisを活性化します。 |
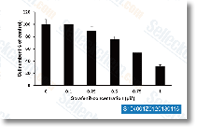
|
|
| S1526 | Quizartinib (AC220) | Quizartinib (AC220)は、MV4-11細胞およびRS4;11細胞においてそれぞれ1.1 nM/4.2 nMのIC50値を示す、Flt3(ITD/WT)に対する第2世代FLT3阻害剤であり、KIT、PDGFRα、PDGFRβ、RET、およびCSF-1Rよりも10倍選択性が高いです。Quizartinib (AC220)は腫瘍細胞のアポトーシスを誘導します。現在、第3相臨床試験が進行中です。 |

|
|
| S1048 | Tozasertib (VX-680, MK-0457) | Tozasertib (VX-680)は、Aurora Aに対するKiappがセルフリーアッセイで0.6 nMのパンAurora阻害剤であり、Aurora B/Aurora Cに対する効力は低く、他の55のキナーゼよりもAurora Aに対して100倍選択的です。唯一の例外は、Kiが30 nMでTozasertibによって阻害されるFms-related tyrosine kinase-3 (FLT-3)とBCR-ABL tyrosine kinaseです。Tozasertibはapoptosisとautophagyを誘導します。フェーズ2。 |
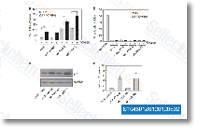
|
|
| S7818 | Pexidartinib (PLX3397) | Pexidartinib (PLX3397)は、CSF-1R、Kit (c-Kit)、およびFLT3を標的とする強力な経口受容体チロシンキナーゼ阻害剤であり、それぞれのIC50値は20 nM、10 nM、および160 nMです。Pexidartinib (PLX3397)は、抗腫瘍活性を伴うアポトーシスと壊死を誘発します。第3相。 |
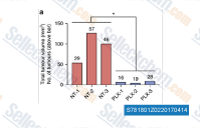
|
|
| S2194 | R406 | R406(R406 besylate)は、無細胞アッセイにおいてIC50が41 nMの強力なSyk阻害剤であり、LynではなくSykを強く阻害し、FLT3に対しては5分の1の効力です。R406はアポトーシスを誘導します。フェーズ1。 |
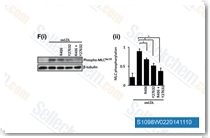
|
|
| S7083 | Ceritinib (LDK378) | Ceritinibは、セルフリーアッセイにおいてIC50が0.2 nMの強力なALK阻害剤です。Ceritinib(LDK378)は、IGF-1R、InsR、STK22D、およびFLT3もそれぞれ8 nM、7 nM、23 nM、および60 nMのIC50で阻害します。第3相。 |
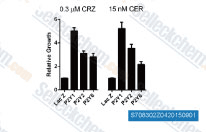
|
|
| S1533 | R406 (free base) | R406 (free base) は、cell-free assay において 41 nM の IC50 を持つ強力な Syk 阻害剤であり、Lyn ではなく Syk を強く阻害しますが、Flt3 に対しては 5 分の 1 の効力です。R406 (free base) はアポトーシスを誘発します。フェーズ 1。 |
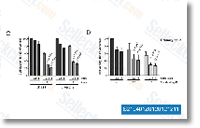
|
|
| S4001 | Cabozantinib S-malate | Cabozantinib malate (XL184)は、強力なVEGFR2阻害剤であるCabozantinibのリンゴ酸塩で、IC50は0.035 nMです。また、細胞フリーアッセイにおいて、c-Met、Ret (c-Ret)、Kit (c-Kit)、Flt-1/3/4、Tie2、AXLをそれぞれIC50が1.3 nM、4 nM、4.6 nM、12 nM/11.3 nM/6 nM、14.3 nM、7 nMで阻害します。Cabozantinib malate (XL184)はアポトーシスを誘発します。 |
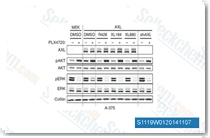
|
|
| S2736 | Fedratinib (TG101348) | Fedratinib (SAR302503, TG101348) は、無細胞アッセイにおいてIC50が3 nMの選択的JAK2阻害剤であり、JAK1およびJAK3に対し、JAK2に対してそれぞれ35倍および334倍選択的です。Fedratinibはまた、FMS-like tyrosine kinase 3 (FLT3)およびRET (c-RET)をそれぞれ15 nMおよび48 nMのIC50で阻害します。Fedratinibは潜在的な抗腫瘍活性を持ちます。Fedratinibは増殖を阻害し、apoptosisを誘導します。第2相。 |
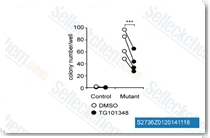
|
|
| S2730 | Crenolanib (CP-868596) | Crenolanibは、CHO細胞において2.1nM/3.2nMのKdを持つPDGFRα/βの強力かつ選択的な阻害剤であり、FLT3も強力に阻害します。D842V変異には感受性を示しますが、V561D変異には示しません。c-Kit、VEGFR-2、TIE-2、FGFR-2、EGFR、erbB2、およびSrcよりもPDGFRに対して100倍以上選択的です。Crenolanibはmitophagyの誘導を助けます。 |
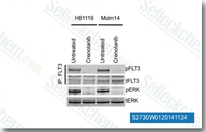
|
|
| S1111 | Foretinib (GSK1363089, XL880) | Foretinibは、HGFRおよびVEGFRのATP競合阻害剤であり、主にMet (c-Met)およびKDRに対して、セルリーアッセイで0.4 nMおよび0.9 nMのIC50を示します。Ron、Flt-1/3/4、Kit (c-Kit)、PDGFRα/β、Tie-2に対しては効力が弱く、FGFR1およびEGFRに対してはほとんど活性がありません。第2相。 |

|
|
| S7754 | ASP2215 (Gilteritinib) | Gilteritinib (ASP2215)は、FLT3およびAXLに対するIC50値がそれぞれ0.29 nMおよび0.73 nMである低分子FLT3/AXL阻害剤です。c-KITを阻害するのに必要な濃度(230 nM)と比較して、FLT3を阻害するIC50値が約800倍強力でした。 | ||
| S1018 | Dovitinib (TKI-258) | Dovitinib (TKI258, CHIR258)は、多標的RTK阻害剤であり、主にクラスIII(FLT3/c-Kit)に対してIC50が1 nM/2 nMであり、クラスIV(FGFR1/3)およびクラスV(VEGFR1-4)RTKに対してもIC50が8-13 nMで強力ですが、セルフリーアッセイではInsR、EGFR、c-Met、EphA2、Tie2、IGF-1R、HER2に対しては効力が低いです。フェーズ4。 |

|
|
| S8032 | PRT062607 (P505-15) HCl | PRT062607 (P505-15, BIIB057, PRT-2607)は、新規で高選択的なSyk阻害剤であり、セルフリーアッセイでのIC50は1 nMで、Fgr、PAK5、Lyn、FAK、Pyk2、FLT3、MLK1、Zap70よりも80倍以上Sykに対して選択性があります。 |
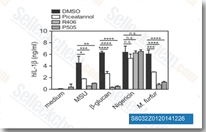
|
|
| S8229 | Brigatinib | Brigatinibは、強力で選択的なALK(IC50、0.6 nM)およびROS1(IC50、0.9 nM)阻害剤です。また、IGF-1R、FLT3、およびFLT3(D835Y)とEGFRの変異型をより低い効力で阻害します。 |

|
|
| S2198 | SGI-1776 free base | SGI-1776 free baseは、ATP競合的な新規のPim1阻害剤であり、セルフリーアッセイではIC50が7 nMで、Pim2およびPim3に対して50倍および10倍の選択性を示し、Flt3およびhaspinにも強力です。SGI-1776はapoptosisおよびautophagyを誘導します。 |

|
|
| S7119 | Go 6976 | Go6976 (PD406976) は、PKC (ラット脳) に対して7.9 nM、PKCα に対して2.3 nM、PKCβ1 に対して6.2 nM のIC50 値を示す強力なPKC阻害剤です。また、JAK2 および Flt3 の強力な阻害剤でもあります。 |

|
|
| S1003 | Linifanib (ABT-869) | Linifanib (ABT-869, AL39324, RG3635) は、KDR、CSF-1R、Flt-1/3、およびPDGFRβに対する新規の強力なATP競合的VEGFR/PDGFR阻害剤であり、それぞれのIC50値は4 nM、3 nM、3 nM/4 nM、および66 nMです。これは主に変異型キナーゼ依存性のがん細胞(すなわちFLT3)に効果的です。Linifanib (ABT-869) はautophagyとapoptosisを誘導します。現在フェーズ3です。 |
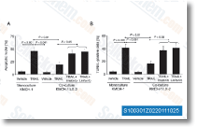
|
|
| S7765 | Dovitinib (TKI258) Lactate monohydrate | Dovitinib (TKI258) Lactate monohydrateはDovitinibの乳酸塩であり、主にクラスIII (FLT3/c-Kit) の多標的RTK阻害剤でIC50は1 nM/2 nMです。また、クラスIV (FGFR1/3) およびクラスV (VEGFR1-4) のRTKに対しても8-13 nMのIC50で強力であり、InsR、EGFR、c-Met、EphA2、Tie2、IGFR1、HER2に対する効力は低いです。第4相。 |
|
|
| S8057 | Pacritinib (SB1518) | Pacritinibは、Janus Kinase 2 (JAK2)およびFms-Like Tyrosine Kinase-3 (FLT3)の強力かつ選択的な阻害剤であり、セル-フリーアッセイにおけるIC50sはそれぞれ23 nMおよび22 nMです。フェーズ3。 |

|
|
| S1244 | Amuvatinib (MP-470) | Amuvatinib (MP-470, HPK 56)は、c-Kit、PDGFRα、およびFlt3の強力な多標的阻害剤であり、それぞれ10 nM、40 nM、81 nMのIC50を示します。Amuvatinibはc-METとc-RETを抑制します。Amuvatinibは、抗腫瘍活性を持つDNA修復タンパク質Rad51阻害剤としても活性があります。フェーズ2。 |

|
|
| S2692 | TG101209 | TG101209は、細胞フリーアッセイにおいて、JAK2に対する選択的阻害剤であり、IC50値は6 nMです。Flt3およびRET (c-RET)に対しては効力が低く、IC50値はそれぞれ25 nMおよび17 nMです。JAK2に対する選択性はJAK3よりも約30倍高く、JAK2V617FおよびMPLW515L/K変異に対して感受性があります。 |
|
|
| S1043 | Tandutinib (MLN518) | Tandutinib (MLN518, CT53518, NSC726292)は、0.22 μMのIC50を持つ強力なFLT3アンタゴニストであり、PDGFRおよびc-Kitも阻害します。FLT3に対するCSF-1Rよりも15〜20倍高い効力があり、FLT3に対するFGFR、EGFR、KDRよりも100倍以上の選択性があります。第2相です。 |

|
|
| S1181 | ENMD-2076 | ENMD-2076は、Aurora AおよびFlt3に対し選択的な活性を持ち、IC50値はそれぞれ14 nMおよび1.86 nMです。Aurora BよりもAurora Aに対して25倍選択的であり、RET、SRC、NTRK1/TRKA、CSF1R/FMS、VEGFR2/KDR、FGFR、PDGFRαに対してはより低効力です。ENMD-2076は、広範囲のヒト固形腫瘍および造血器癌細胞株の増殖を0.025〜0.7 μMのIC50で阻害し、apoptosisおよびG2/M期停止を誘導します。フェーズ2。 |

|
|
| S2158 | KW-2449 | KW-2449は、IC50が6.6 nMで主にFLT3を標的とする多標的阻害剤であり、FGFR1、Bcr-Abl、Aurora Aに対して中程度の効力があります。PDGFRβ、IGF-1R、EGFRにはほとんど影響がありません。第1相。 |

|
|
| S7576 | UNC2025 HCl | UNC2025 HClは、強力で経口投与可能なデュアルMER/FLT3阻害剤であり、IC50はそれぞれ0.74 nMと0.8 nMで、AxlとTyro3に対して約20倍の選択性を示します。 | ||
| S7014 | Merestinib (LY2801653) | Merestinib (LY2801653)は、解離定数(Ki)が2 nM、薬力学的滞留時間(Koff)が0.00132 min(-1)、t1/2が525 minの、Met (c-Met)チロシンキナーゼに対するタイプII ATP競合性スローオフ阻害剤です。Merestinib (LY2801653)は、MST1R、AXL、ROS1、MKNK1/2、FLT3、MERTK、DDR1、DDR2もそれぞれIC50が11 nM、2 nM、23 nM、7 nM、7 nM、10 nM、0.1 nM、7 nMで阻害します。 | ||
| S8442 | TAK-659 Hydrochloride | TAK-659 Hydrochlorideは、3.2 nMのIC50値を持つ、脾臓チロシンキナーゼ(SYK)の強力かつ選択的な阻害剤です。ほとんどの他のキナーゼに対して選択的ですが、SYKとFLT3の両方に対して強力です。 | ||
| S9662 | UNC2025 | UNC2025は、強力で経口活性のあるFLT3およびMERのデュアル阻害剤であり、それぞれIC50は0.35 nMおよび0.46 nMです。UNC2025はまた、AXL、TRKA、TRKC、QIK、TYRO3、SLK、NuaK1、Kit (c-Kit)、およびMet (c-Met)も阻害し、それぞれのIC50は1.65 nM、1.67 nM、4.38 nM、5.75 nM、5.83 nM、6.14 nM、7.97 nM、8.18 nM、および364 nMです。 | ||
| S7003 | AZD2932 | AZD2932は、VEGFR-2、PDGFRβ、Flt-3、およびc-Kitに対してそれぞれ8 nM、4 nM、7 nM、および9 nMのIC50を持つ、強力かつ多標的のプロテインチロシンキナーゼ阻害剤です。 | ||
| S0278 | SU5614 | SU5614 (Chloro-SU5416, Chloro-Semaxanib)は、VEGFR-2、c-kit、および野生型と変異型の両方のFLT3の低分子受容体型チロシンキナーゼ(RTK)阻害剤です。SU5614は細胞増殖を減少させ、アポトーシスを誘発します。 | ||
| S6662 | AST-487 (NVP-AST487) | N,N'-ジフェニル尿素であるAST-487 (NVP-AST487)は、kiが0.12 μMのFlt3のATP競合阻害剤です。FLT3の他に、AST487はRET、KDR、c-KIT、およびc-ABLキナーゼも1 μM以下のIC50値で阻害します。 | ||
| S8023 | TCS 359 | TCS 359は、42 nMのIC50を持つ強力なFLT3阻害剤です。 | ||
| S2018 | ENMD-2076 L-(+)-Tartaric acid | ENMD-2076 L-(+)-酒石酸は、ENMD-2076の酒石酸であり、Aurora AおよびFlt3に対し、IC50が14 nMおよび1.86 nMの選択的活性を示し、Aurora BよりもAurora Aに対して25倍選択的であり、VEGFR2/KDR、VEGFR3、FGFR1、FGFR2、PDGFRαに対する効力は低い。第2相。 |
|
|
| S6839 | MRX-2843 | MRX-2843(UNC2371)は、チロシンキナーゼMERTKとFLT3の経口活性デュアル阻害剤であり、IC50値はそれぞれ1.3 nMと0.64 nMです。 | ||
| S7545 | G-749 | G-749は、FLT3(WT)、FLT3(D835Y)、Merに対してそれぞれ0.4 nM、0.6 nM、1 nMのIC50を示す新規かつ強力なFLT3阻害剤であり、他のチロシンキナーゼに対してはより低い効力を示します。 | ||
| E2387 | FLT3-IN-2 | FLT3-IN-2は、IC50が1 μM未満のFLT3阻害剤です。 | ||
| S9779 | Emavusertib (CA-4948) | Emavusertib (CA-4948)は、抗腫瘍活性を持つインターロイキン-1受容体関連キナーゼ4/FMS様チロシンキナーゼ3 (IRAK4/FLT3)の強力な経口生物学的利用能阻害剤です。 | ||
| S1099 | SKLB4771 (FLT3-IN-1) | SKLB4771 は、IC50 が 10 nM のヒト受容体型チロシンプロテインキナーゼ FLT3 の強力かつ選択的阻害剤です。 | ||
| S7533 | AMG 925 | AMG 925は、強力な経口バイオアベイラビリティを有するデュアルFLT3/CDK4阻害剤であり、IC50はそれぞれ1 nMと3 nMです。 | ||
| S7002 | Zotiraciclib | Zotiraciclib (SB1317; TG02)は、CDK (CDK1、2、9に対するIC50はそれぞれ9、5、3 nM)、FLT3 (IC50 = 19 nM)、およびJAK2 (IC50 = 19 nM)に対して強力な効力を持つ新規低分子強力CDK/JAK2/FLT3阻害剤です。 | ||
| E1437 | PHI-101 | PHI-101は、複数の薬剤耐性変異に対する抵抗性を克服する、経口活性のある強力な第三世代FLT3阻害剤です。再発または難治性の急性骨髄性白血病(AML)の研究に役立つ可能性があります。 | ||
| S0550 | 5'-Fluoroindirubinoxime | インジルビン誘導体である5'-Fluoroindirubinoxime(5’-FIO、化合物13)は、強力なFLT3阻害剤であり、IC50は15 nMです。 | ||
| E4598 | MEN1703(SEL24,SEL24-B489) | MEN1703(SEL24-B489,SEL24, Pim/flt3 kinase inhibitor SEL24)は、PIMおよびFLT3-ITDの強力なタイプIの経口活性デュアル阻害剤であり、それぞれPIM1に対して2nM、PIM2に対して2nM、PIM3に対して3nMのKd値を示します。MEN1703は、急性骨髄性白血病の治療において幅広い治療の可能性を示します。 | ||
| S9275 | Isoguanosine | Isoguanosine(クロトノシド)は、AML治療のためにFLT3およびHDAC3/6を阻害します。Isoguanosineは、クロトン属の種子に見られるグアノシンの天然に存在する活性異性体です。 | ||
| S7640 | Lestaurtinib | Lestaurtinib (CEP-701, KT-5555) は、インドロカルバゾールK252の誘導体であり、スタウロスポリンアナログです。これは、受容体タンパク質チロシンキナーゼ(RPTKs)の経口活性阻害剤であり、FLT3およびTrkをそれぞれ3 nMおよび< 25 nMのIC50で阻害します。また、JAK2を0.9 nMのIC50で阻害します。 | ||
| S5986 | FLT3-IN-3 | FLT3-IN-3は、FLT3 WTおよびFLT3 D835Yに対してそれぞれ13 nMおよび8 nMのIC50値を持つ強力なFLT3阻害剤です。FLT3-IN-3は、低ナノモル濃度でFLT3-ITD陽性のMV4-11およびMOLM-13細胞株の増殖を効果的に抑制します。 | ||
| E4720 | Flonoltinib | Flonoltinib (JAK2/FLT3-IN-1) は、JAK2とFLT3の強力かつ選択的なデュアル阻害剤であり、in vitroキナーゼアッセイにおいて、JAK2、JAK2V617F、およびFLT3に対してそれぞれ0.8、1.4、および15 nMのIC50値を示します。また、血球貪食性リンパ組織球症 (HLH) 患者のCD8+T細胞、CTLL2細胞、およびRAW 264.7細胞において、細胞増殖を阻害し、G2/M期細胞周期停止およびアポトーシスを誘導します。 | ||
| S6060 | HPK1-IN-2 | HPK1-IN-2は、造血前駆細胞キナーゼ-1(HPK1、セリン/スレオニンSte20関連プロテインキナーゼ)、Lck、およびFlt3をそれぞれIC50 <0.05 μM、IC50 <0.5 μM、IC50 <0.05 μMで阻害する、強力で経口活性のある阻害剤です。HPK1-IN-2は抗腫瘍活性を示します。 | ||
| E2658 | FN-1501 | FN-1501は、強力なFms様受容体型チロシンキナーゼ3 (FLT3)およびサイクリン依存性キナーゼ (CDK)阻害剤であり、CDK2/サイクリン A、CDK4/サイクリン D1、CDK6/サイクリン D1、およびFLT3に対するIC50値はそれぞれ2.47、0.85、1.96、0.28 nMです。 | ||
| S9892 | HM43239 | HM43239 は、FLT3 野生型、ITD 変異体、TKD 変異体だけでなく、FLT3 ITD/TKD 二重変異も選択的に阻害する、新規の強力な低分子 FLT3 阻害剤です。FLT3 WT、FLT3 ITD、FLT3 D835Y キナーゼに対する HM43239 の IC50 は、それぞれ 1.1 nM、1.8 nM、1.0 nM です。 | ||
| S1460 | SP600125 | SP600125(Nsc75890)は、JNK1、JNK2、JNK3に対する広範なJNK阻害剤であり、細胞フリーアッセイでのIC50はそれぞれ40nM、40nM、90nMです。MKK4に対して10倍、MKK3、MKK6、PKB、PKCαに対して25倍、ERK2、p38、Chk1、EGFRなどに対して100倍高い選択性を示します。SP600125は、Aurora kinase A、FLT3、TRKAを含むserine/threonine kinasesの広範な阻害剤でもあり、IC50はそれぞれ60nM、90nM、70nMです。SP600125はautophagyを阻害し、apoptosisを活性化します。 |

|
|
| S7397 | Sorafenib (BAY 43-9006) | Sorafenib は、細胞を含まないアッセイで Raf-1 および B-Raf のマルチキナーゼ阻害剤であり、それぞれ 6 nM および 22 nM の IC50 を示します。Sorafenib は、VEGFR-2、VEGFR-3、PDGFR-β、Flt-3 および c-KIT をそれぞれ 90 nM、20 nM、57 nM、59 nM および 68 nM の IC50 で阻害します。Sorafenib は、抗腫瘍活性を持つ autophagy と apoptosis を誘導し、ferroptosis を活性化します。 |

|
|
| S1040 | Sorafenib Tosylate (BAY 43-9006) | Sorafenib tosylateは、無細胞アッセイにおいてIC50がそれぞれ6 nMおよび22 nMのRaf-1およびB-Rafのマルチキナーゼ阻害剤です。Sorafenib Tosylateは、VEGFR-2、VEGFR-3、PDGFR-β、Flt-3、およびc-KITをそれぞれ90 nM、20 nM、57 nM、59 nM、および68 nMのIC50で阻害します。Sorafenib Tosylateは、抗腫瘍活性を伴うautophagyとapoptosisを誘導し、ferroptosisを活性化します。 |

|
|
| S1526 | Quizartinib (AC220) | Quizartinib (AC220)は、MV4-11細胞およびRS4;11細胞においてそれぞれ1.1 nM/4.2 nMのIC50値を示す、Flt3(ITD/WT)に対する第2世代FLT3阻害剤であり、KIT、PDGFRα、PDGFRβ、RET、およびCSF-1Rよりも10倍選択性が高いです。Quizartinib (AC220)は腫瘍細胞のアポトーシスを誘導します。現在、第3相臨床試験が進行中です。 |

|
|
| S1048 | Tozasertib (VX-680, MK-0457) | Tozasertib (VX-680)は、Aurora Aに対するKiappがセルフリーアッセイで0.6 nMのパンAurora阻害剤であり、Aurora B/Aurora Cに対する効力は低く、他の55のキナーゼよりもAurora Aに対して100倍選択的です。唯一の例外は、Kiが30 nMでTozasertibによって阻害されるFms-related tyrosine kinase-3 (FLT-3)とBCR-ABL tyrosine kinaseです。Tozasertibはapoptosisとautophagyを誘導します。フェーズ2。 |

|
|
| S7818 | Pexidartinib (PLX3397) | Pexidartinib (PLX3397)は、CSF-1R、Kit (c-Kit)、およびFLT3を標的とする強力な経口受容体チロシンキナーゼ阻害剤であり、それぞれのIC50値は20 nM、10 nM、および160 nMです。Pexidartinib (PLX3397)は、抗腫瘍活性を伴うアポトーシスと壊死を誘発します。第3相。 |

|
|
| S2194 | R406 | R406(R406 besylate)は、無細胞アッセイにおいてIC50が41 nMの強力なSyk阻害剤であり、LynではなくSykを強く阻害し、FLT3に対しては5分の1の効力です。R406はアポトーシスを誘導します。フェーズ1。 |

|
|
| S7083 | Ceritinib (LDK378) | Ceritinibは、セルフリーアッセイにおいてIC50が0.2 nMの強力なALK阻害剤です。Ceritinib(LDK378)は、IGF-1R、InsR、STK22D、およびFLT3もそれぞれ8 nM、7 nM、23 nM、および60 nMのIC50で阻害します。第3相。 |

|
|
| S1533 | R406 (free base) | R406 (free base) は、cell-free assay において 41 nM の IC50 を持つ強力な Syk 阻害剤であり、Lyn ではなく Syk を強く阻害しますが、Flt3 に対しては 5 分の 1 の効力です。R406 (free base) はアポトーシスを誘発します。フェーズ 1。 |

|
|
| S4001 | Cabozantinib S-malate | Cabozantinib malate (XL184)は、強力なVEGFR2阻害剤であるCabozantinibのリンゴ酸塩で、IC50は0.035 nMです。また、細胞フリーアッセイにおいて、c-Met、Ret (c-Ret)、Kit (c-Kit)、Flt-1/3/4、Tie2、AXLをそれぞれIC50が1.3 nM、4 nM、4.6 nM、12 nM/11.3 nM/6 nM、14.3 nM、7 nMで阻害します。Cabozantinib malate (XL184)はアポトーシスを誘発します。 |

|
|
| S2736 | Fedratinib (TG101348) | Fedratinib (SAR302503, TG101348) は、無細胞アッセイにおいてIC50が3 nMの選択的JAK2阻害剤であり、JAK1およびJAK3に対し、JAK2に対してそれぞれ35倍および334倍選択的です。Fedratinibはまた、FMS-like tyrosine kinase 3 (FLT3)およびRET (c-RET)をそれぞれ15 nMおよび48 nMのIC50で阻害します。Fedratinibは潜在的な抗腫瘍活性を持ちます。Fedratinibは増殖を阻害し、apoptosisを誘導します。第2相。 |

|
|
| S2730 | Crenolanib (CP-868596) | Crenolanibは、CHO細胞において2.1nM/3.2nMのKdを持つPDGFRα/βの強力かつ選択的な阻害剤であり、FLT3も強力に阻害します。D842V変異には感受性を示しますが、V561D変異には示しません。c-Kit、VEGFR-2、TIE-2、FGFR-2、EGFR、erbB2、およびSrcよりもPDGFRに対して100倍以上選択的です。Crenolanibはmitophagyの誘導を助けます。 |

|
|
| S1111 | Foretinib (GSK1363089, XL880) | Foretinibは、HGFRおよびVEGFRのATP競合阻害剤であり、主にMet (c-Met)およびKDRに対して、セルリーアッセイで0.4 nMおよび0.9 nMのIC50を示します。Ron、Flt-1/3/4、Kit (c-Kit)、PDGFRα/β、Tie-2に対しては効力が弱く、FGFR1およびEGFRに対してはほとんど活性がありません。第2相。 |

|
|
| S7754 | ASP2215 (Gilteritinib) | Gilteritinib (ASP2215)は、FLT3およびAXLに対するIC50値がそれぞれ0.29 nMおよび0.73 nMである低分子FLT3/AXL阻害剤です。c-KITを阻害するのに必要な濃度(230 nM)と比較して、FLT3を阻害するIC50値が約800倍強力でした。 | ||
| S1018 | Dovitinib (TKI-258) | Dovitinib (TKI258, CHIR258)は、多標的RTK阻害剤であり、主にクラスIII(FLT3/c-Kit)に対してIC50が1 nM/2 nMであり、クラスIV(FGFR1/3)およびクラスV(VEGFR1-4)RTKに対してもIC50が8-13 nMで強力ですが、セルフリーアッセイではInsR、EGFR、c-Met、EphA2、Tie2、IGF-1R、HER2に対しては効力が低いです。フェーズ4。 |

|
|
| S8032 | PRT062607 (P505-15) HCl | PRT062607 (P505-15, BIIB057, PRT-2607)は、新規で高選択的なSyk阻害剤であり、セルフリーアッセイでのIC50は1 nMで、Fgr、PAK5、Lyn、FAK、Pyk2、FLT3、MLK1、Zap70よりも80倍以上Sykに対して選択性があります。 |

|
|
| S8229 | Brigatinib | Brigatinibは、強力で選択的なALK(IC50、0.6 nM)およびROS1(IC50、0.9 nM)阻害剤です。また、IGF-1R、FLT3、およびFLT3(D835Y)とEGFRの変異型をより低い効力で阻害します。 |

|
|
| S2198 | SGI-1776 free base | SGI-1776 free baseは、ATP競合的な新規のPim1阻害剤であり、セルフリーアッセイではIC50が7 nMで、Pim2およびPim3に対して50倍および10倍の選択性を示し、Flt3およびhaspinにも強力です。SGI-1776はapoptosisおよびautophagyを誘導します。 |

|
|
| S7119 | Go 6976 | Go6976 (PD406976) は、PKC (ラット脳) に対して7.9 nM、PKCα に対して2.3 nM、PKCβ1 に対して6.2 nM のIC50 値を示す強力なPKC阻害剤です。また、JAK2 および Flt3 の強力な阻害剤でもあります。 |

|
|
| S1003 | Linifanib (ABT-869) | Linifanib (ABT-869, AL39324, RG3635) は、KDR、CSF-1R、Flt-1/3、およびPDGFRβに対する新規の強力なATP競合的VEGFR/PDGFR阻害剤であり、それぞれのIC50値は4 nM、3 nM、3 nM/4 nM、および66 nMです。これは主に変異型キナーゼ依存性のがん細胞(すなわちFLT3)に効果的です。Linifanib (ABT-869) はautophagyとapoptosisを誘導します。現在フェーズ3です。 |

|
|
| S7765 | Dovitinib (TKI258) Lactate monohydrate | Dovitinib (TKI258) Lactate monohydrateはDovitinibの乳酸塩であり、主にクラスIII (FLT3/c-Kit) の多標的RTK阻害剤でIC50は1 nM/2 nMです。また、クラスIV (FGFR1/3) およびクラスV (VEGFR1-4) のRTKに対しても8-13 nMのIC50で強力であり、InsR、EGFR、c-Met、EphA2、Tie2、IGFR1、HER2に対する効力は低いです。第4相。 |

|
|
| S8057 | Pacritinib (SB1518) | Pacritinibは、Janus Kinase 2 (JAK2)およびFms-Like Tyrosine Kinase-3 (FLT3)の強力かつ選択的な阻害剤であり、セル-フリーアッセイにおけるIC50sはそれぞれ23 nMおよび22 nMです。フェーズ3。 |

|
|
| S1244 | Amuvatinib (MP-470) | Amuvatinib (MP-470, HPK 56)は、c-Kit、PDGFRα、およびFlt3の強力な多標的阻害剤であり、それぞれ10 nM、40 nM、81 nMのIC50を示します。Amuvatinibはc-METとc-RETを抑制します。Amuvatinibは、抗腫瘍活性を持つDNA修復タンパク質Rad51阻害剤としても活性があります。フェーズ2。 |

|
|
| S2692 | TG101209 | TG101209は、細胞フリーアッセイにおいて、JAK2に対する選択的阻害剤であり、IC50値は6 nMです。Flt3およびRET (c-RET)に対しては効力が低く、IC50値はそれぞれ25 nMおよび17 nMです。JAK2に対する選択性はJAK3よりも約30倍高く、JAK2V617FおよびMPLW515L/K変異に対して感受性があります。 |

|
|
| S1043 | Tandutinib (MLN518) | Tandutinib (MLN518, CT53518, NSC726292)は、0.22 μMのIC50を持つ強力なFLT3アンタゴニストであり、PDGFRおよびc-Kitも阻害します。FLT3に対するCSF-1Rよりも15〜20倍高い効力があり、FLT3に対するFGFR、EGFR、KDRよりも100倍以上の選択性があります。第2相です。 |

|
|
| S1181 | ENMD-2076 | ENMD-2076は、Aurora AおよびFlt3に対し選択的な活性を持ち、IC50値はそれぞれ14 nMおよび1.86 nMです。Aurora BよりもAurora Aに対して25倍選択的であり、RET、SRC、NTRK1/TRKA、CSF1R/FMS、VEGFR2/KDR、FGFR、PDGFRαに対してはより低効力です。ENMD-2076は、広範囲のヒト固形腫瘍および造血器癌細胞株の増殖を0.025〜0.7 μMのIC50で阻害し、apoptosisおよびG2/M期停止を誘導します。フェーズ2。 |

|
|
| S2158 | KW-2449 | KW-2449は、IC50が6.6 nMで主にFLT3を標的とする多標的阻害剤であり、FGFR1、Bcr-Abl、Aurora Aに対して中程度の効力があります。PDGFRβ、IGF-1R、EGFRにはほとんど影響がありません。第1相。 |

|
|
| S7576 | UNC2025 HCl | UNC2025 HClは、強力で経口投与可能なデュアルMER/FLT3阻害剤であり、IC50はそれぞれ0.74 nMと0.8 nMで、AxlとTyro3に対して約20倍の選択性を示します。 | ||
| S7014 | Merestinib (LY2801653) | Merestinib (LY2801653)は、解離定数(Ki)が2 nM、薬力学的滞留時間(Koff)が0.00132 min(-1)、t1/2が525 minの、Met (c-Met)チロシンキナーゼに対するタイプII ATP競合性スローオフ阻害剤です。Merestinib (LY2801653)は、MST1R、AXL、ROS1、MKNK1/2、FLT3、MERTK、DDR1、DDR2もそれぞれIC50が11 nM、2 nM、23 nM、7 nM、7 nM、10 nM、0.1 nM、7 nMで阻害します。 | ||
| S8442 | TAK-659 Hydrochloride | TAK-659 Hydrochlorideは、3.2 nMのIC50値を持つ、脾臓チロシンキナーゼ(SYK)の強力かつ選択的な阻害剤です。ほとんどの他のキナーゼに対して選択的ですが、SYKとFLT3の両方に対して強力です。 | ||
| S9662 | UNC2025 | UNC2025は、強力で経口活性のあるFLT3およびMERのデュアル阻害剤であり、それぞれIC50は0.35 nMおよび0.46 nMです。UNC2025はまた、AXL、TRKA、TRKC、QIK、TYRO3、SLK、NuaK1、Kit (c-Kit)、およびMet (c-Met)も阻害し、それぞれのIC50は1.65 nM、1.67 nM、4.38 nM、5.75 nM、5.83 nM、6.14 nM、7.97 nM、8.18 nM、および364 nMです。 | ||
| S7003 | AZD2932 | AZD2932は、VEGFR-2、PDGFRβ、Flt-3、およびc-Kitに対してそれぞれ8 nM、4 nM、7 nM、および9 nMのIC50を持つ、強力かつ多標的のプロテインチロシンキナーゼ阻害剤です。 | ||
| S0278 | SU5614 | SU5614 (Chloro-SU5416, Chloro-Semaxanib)は、VEGFR-2、c-kit、および野生型と変異型の両方のFLT3の低分子受容体型チロシンキナーゼ(RTK)阻害剤です。SU5614は細胞増殖を減少させ、アポトーシスを誘発します。 | ||
| S6662 | AST-487 (NVP-AST487) | N,N'-ジフェニル尿素であるAST-487 (NVP-AST487)は、kiが0.12 μMのFlt3のATP競合阻害剤です。FLT3の他に、AST487はRET、KDR、c-KIT、およびc-ABLキナーゼも1 μM以下のIC50値で阻害します。 | ||
| S8023 | TCS 359 | TCS 359は、42 nMのIC50を持つ強力なFLT3阻害剤です。 | ||
| S2018 | ENMD-2076 L-(+)-Tartaric acid | ENMD-2076 L-(+)-酒石酸は、ENMD-2076の酒石酸であり、Aurora AおよびFlt3に対し、IC50が14 nMおよび1.86 nMの選択的活性を示し、Aurora BよりもAurora Aに対して25倍選択的であり、VEGFR2/KDR、VEGFR3、FGFR1、FGFR2、PDGFRαに対する効力は低い。第2相。 |
|
|
| S6839 | MRX-2843 | MRX-2843(UNC2371)は、チロシンキナーゼMERTKとFLT3の経口活性デュアル阻害剤であり、IC50値はそれぞれ1.3 nMと0.64 nMです。 | ||
| S7545 | G-749 | G-749は、FLT3(WT)、FLT3(D835Y)、Merに対してそれぞれ0.4 nM、0.6 nM、1 nMのIC50を示す新規かつ強力なFLT3阻害剤であり、他のチロシンキナーゼに対してはより低い効力を示します。 | ||
| E2387 | FLT3-IN-2 | FLT3-IN-2は、IC50が1 μM未満のFLT3阻害剤です。 | ||
| S9779 | Emavusertib (CA-4948) | Emavusertib (CA-4948)は、抗腫瘍活性を持つインターロイキン-1受容体関連キナーゼ4/FMS様チロシンキナーゼ3 (IRAK4/FLT3)の強力な経口生物学的利用能阻害剤です。 | ||
| S1099 | SKLB4771 (FLT3-IN-1) | SKLB4771 は、IC50 が 10 nM のヒト受容体型チロシンプロテインキナーゼ FLT3 の強力かつ選択的阻害剤です。 | ||
| S7533 | AMG 925 | AMG 925は、強力な経口バイオアベイラビリティを有するデュアルFLT3/CDK4阻害剤であり、IC50はそれぞれ1 nMと3 nMです。 | ||
| S7002 | Zotiraciclib | Zotiraciclib (SB1317; TG02)は、CDK (CDK1、2、9に対するIC50はそれぞれ9、5、3 nM)、FLT3 (IC50 = 19 nM)、およびJAK2 (IC50 = 19 nM)に対して強力な効力を持つ新規低分子強力CDK/JAK2/FLT3阻害剤です。 | ||
| E1437 | PHI-101 | PHI-101は、複数の薬剤耐性変異に対する抵抗性を克服する、経口活性のある強力な第三世代FLT3阻害剤です。再発または難治性の急性骨髄性白血病(AML)の研究に役立つ可能性があります。 | ||
| S0550 | 5'-Fluoroindirubinoxime | インジルビン誘導体である5'-Fluoroindirubinoxime(5’-FIO、化合物13)は、強力なFLT3阻害剤であり、IC50は15 nMです。 | ||
| E4598 | MEN1703(SEL24,SEL24-B489) | MEN1703(SEL24-B489,SEL24, Pim/flt3 kinase inhibitor SEL24)は、PIMおよびFLT3-ITDの強力なタイプIの経口活性デュアル阻害剤であり、それぞれPIM1に対して2nM、PIM2に対して2nM、PIM3に対して3nMのKd値を示します。MEN1703は、急性骨髄性白血病の治療において幅広い治療の可能性を示します。 | ||
| S9275 | Isoguanosine | Isoguanosine(クロトノシド)は、AML治療のためにFLT3およびHDAC3/6を阻害します。Isoguanosineは、クロトン属の種子に見られるグアノシンの天然に存在する活性異性体です。 | ||
| S7640 | Lestaurtinib | Lestaurtinib (CEP-701, KT-5555) は、インドロカルバゾールK252の誘導体であり、スタウロスポリンアナログです。これは、受容体タンパク質チロシンキナーゼ(RPTKs)の経口活性阻害剤であり、FLT3およびTrkをそれぞれ3 nMおよび< 25 nMのIC50で阻害します。また、JAK2を0.9 nMのIC50で阻害します。 | ||
| S5986 | FLT3-IN-3 | FLT3-IN-3は、FLT3 WTおよびFLT3 D835Yに対してそれぞれ13 nMおよび8 nMのIC50値を持つ強力なFLT3阻害剤です。FLT3-IN-3は、低ナノモル濃度でFLT3-ITD陽性のMV4-11およびMOLM-13細胞株の増殖を効果的に抑制します。 | ||
| E4720 | Flonoltinib | Flonoltinib (JAK2/FLT3-IN-1) は、JAK2とFLT3の強力かつ選択的なデュアル阻害剤であり、in vitroキナーゼアッセイにおいて、JAK2、JAK2V617F、およびFLT3に対してそれぞれ0.8、1.4、および15 nMのIC50値を示します。また、血球貪食性リンパ組織球症 (HLH) 患者のCD8+T細胞、CTLL2細胞、およびRAW 264.7細胞において、細胞増殖を阻害し、G2/M期細胞周期停止およびアポトーシスを誘導します。 | ||
| S6060 | HPK1-IN-2 | HPK1-IN-2は、造血前駆細胞キナーゼ-1(HPK1、セリン/スレオニンSte20関連プロテインキナーゼ)、Lck、およびFlt3をそれぞれIC50 <0.05 μM、IC50 <0.5 μM、IC50 <0.05 μMで阻害する、強力で経口活性のある阻害剤です。HPK1-IN-2は抗腫瘍活性を示します。 | ||
| E2658 | FN-1501 | FN-1501は、強力なFms様受容体型チロシンキナーゼ3 (FLT3)およびサイクリン依存性キナーゼ (CDK)阻害剤であり、CDK2/サイクリン A、CDK4/サイクリン D1、CDK6/サイクリン D1、およびFLT3に対するIC50値はそれぞれ2.47、0.85、1.96、0.28 nMです。 | ||
| S9892 | HM43239 | HM43239 は、FLT3 野生型、ITD 変異体、TKD 変異体だけでなく、FLT3 ITD/TKD 二重変異も選択的に阻害する、新規の強力な低分子 FLT3 阻害剤です。FLT3 WT、FLT3 ITD、FLT3 D835Y キナーゼに対する HM43239 の IC50 は、それぞれ 1.1 nM、1.8 nM、1.0 nM です。 |
FLT3シグナル伝達経路
納期 国内在庫品:受注日の翌日(15時までの受注分) *北海道、九州、沖縄への配送は受注日より2日以上 を要する場合あり 海外在庫品:受注後1〜2週間